Animal models of Parkinson's disease
Animal models of Parkinson's disease are essential in the research field and widely used to study Parkinson's disease. Parkinson's disease is a neurodegenerative disorder, characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc). The loss of the dopamine neurons in the brain, results in motor dysfunction, ultimately causing the four cardinal symptoms of PD: tremor, rigidity, postural instability, and bradykinesia.[1] It is the second most prevalent neurodegenerative disease, following Alzheimer's disease. It is estimated that nearly one million people could be living with PD in the United States.[2]
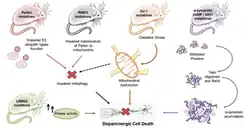
There are a variety of models that can be utilized to be able to address important aspects of Parkinson's disease. Researchers can consider disease progression, cell death, behavioral characteristics, and more PD phenotypes. Parkinson's disease animal models are divided into two categories: neurotoxin models and genetic models.[3] Neurotoxin models include chemically induced toxicity in the brain; whereas, genetic models include genes that are mutated and induce PD phenotypes.
Neurotoxin models
6-OHDA
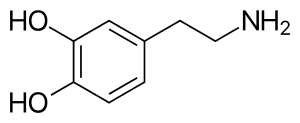
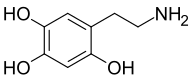
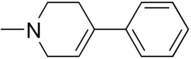
6-Hydroxydopamine, better known as 6-OHDA, is a widely used neurotoxin in PD models. It is structurally similar to dopamine, only differentiating by an additional hydroxyl group in the 6-OHDA structure (Figure 1 & Figure 2).[4] Through scientific studies, this neurotoxin has been used in rodents (rats and mice), guinea pigs, cats, dogs, and monkeys. 6-OHDA does not cross the blood-brain-barrier (BBB) making the chemical more selective for dopaminergic neurons. This model requires injecting the 6-OHDA directly into the nigrostriatal pathway, targeting the dopamine transporter (DAT).This can be performed through stereotaxic injections (both unilateral and bilateral are experimentally permissible) and will eventually cause loss of dopamine neurons in the SNpc and loss of dopamine terminals in the striatum since the nigrostriatal pathway is being affected.[5][4][6] The neurotoxin can be injected has been shown to be injected into the striatum and the substantia nigra. However, injections into the SNpc is estimated to degrade about 60% of tyrosine hydroxylase (TH+) neurons as well as loss of TH positive terminals in the striatum. A limitation to using 6-OHDA is that the potency of the neurotoxin causes rapid apoptosis, which makes it difficult to study Parkinson's disease progression.[5]
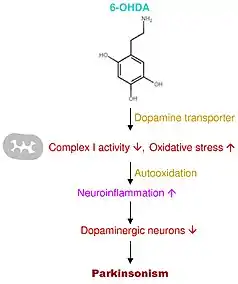
The mechanism of action of 6-OHDA occurs through the aggregation of toxins and the conversion into catecholaminergic neurons. Since the structures of both dopamine and 6-OHDA are similar, the dopamine transporter takes up the 6-OHDA and induces toxicity.[7][6] This toxicity emerges from the production of free radicals from the additional hydroxyl group in the neurotoxin's structure. There is also oxidative stress occurring mediated through the inhibition of the cell's mitochondrial complex I, producing ROS (reactive oxygen species), which causes a decrease or loss in respiratory activity. In addition, there is also the proposed mechanism of oxidative stress inducing neuroinflammation (Figure 3).[4][6]

MPTP
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is a widely used neurotoxin in Parkinson's disease research (Figure 4). In contrast to 6-OHDA, MPTP crosses the BBB which making the neurotoxin even more selective for dopaminergic neurons. Due to the ability to cross the blood-brain-barrier, MPTP is administered peripherally, subcutaneously. This neurotoxin is known to replicate oxidative stress, ROS, energy failure, and inflammation; which are all hallmarks in Parkinson's disease. However, it does not produce Lewy body pathology. The mechanism of action of MPTP is due to its conversion to 1-methyl-4-phenylpyridinium (MPP+) caused by the interaction of MPTP with monoamine oxidase B (MAO-B). (Figure5). MPTP enters astrocytes and is metabolized to MPP+ before being released. Once released into the extracellular space, MPP+ is taken up into the neuron by DAT and is stored in vesicles by the up take of vesicular monoamine transporter (VMAT2). In the neuron, MPP+ inhibits the function of complex 1 of electron transport chain, which decreases ATP production and releases ROS.
Herbicides (rotenone and paraquat)
Rotenone is a chemical compound (Figure 6) that can be derived from the plants: Derris elliptica, D mallaccensis, Lonchocarpus utilis, and L urucu.[8] It is a known neurotoxin that is selective to dopaminergic neurons when administered to rodents via stereotaxic injections. However, it targets the striatum and not the substantia nigra. Moreover, rotenone can cross the BBB and spread through the central nervous system.[9][10] Since rotenone can cross the blood-brain-barrier, it can be administered peripherally as well. Although, peripheral injections can lead to system toxicity.[11] The exact mechanism of action of rotenone is still unclear, but one aspect that is known is that the herbicide accumulates and clusters in the neuron in organelles like the mitochondria, which disrupts the oxidative phosphorylation mechanism in the cell and inhibits the respiratory chain complex I. Limitations of using rotenone is the lack of reproducibility of results throughout experiments and the quantity of aggregates and lesion. In addition, there is an elevated mortality rate in the animals induced with rotenone.[10][9]
Paraquat
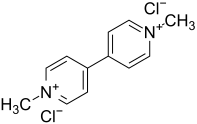
1,1'-dimethyl-4-4'-bipyridinium dichloride (Paraquat) is a nonselective herbicide (Figure 7).[12] Human exposure to this chemical is highly toxic. Its chemical structure is very similar to MPP+, therefore, it was thought to act as a neurotoxin as well.[4] Paraquat has the capability to cross the BBB and is selective to dopamine neurons when injected via stereotaxic injections in the brain. Similar to rotenone, paraquat can also be administered peripherally, however, this can lead to systemic toxicity.[11] It is found to decrease dopamine concentration and produce parkinsonian phenotypes (both physically and behaviorally). Mechanistically, paraquat targets the dopamine transporter to be transported into dopaminergic neurons and ends up in the striatum. It lingers in the midbrain for approximately four weeks. However, since it is capable to cross the BBB, the toxin can be found in other regions such as the pineal gland, cerebral ventricles, olfactory bulb, hypothalamus, and the area postrema. Several studies have demonstrated the relationship between paraquat and oxidative stress indicating that this may be another mechanism of paraquat induced neurodegeneration. In addition, the herbicide is accumulated in the lungs and kidney, resulting high toxicity; as well, as death.[9][10]
Genetic models
alpha-synuclein
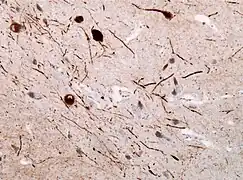
Alpha-synuclein (α-synuclein) is an endogenous protein that is encoded by the SNCA gene and known as the pathological hallmark of Parkinson's disease.[13] It is found in distinct regions of the body, but in PD, alpha-synuclein accumulation in the brain is of main importance. This protein misfolds and accumulates creating insoluble aggregates in the brain known as Lewy bodies (found in the soma) and Lewy neurites (found in the neuropil)(Figure 8). This pathology is well known as synucleinopathies.[8][13] The inclusions/aggregates lead to dopamine neuronal depletion in the SNpc as well as dopamine terminal loss in the striatum from the projection of SNpc neurons through the nigrostriatal pathway. In addition, studies have shown that there is progressive formation of α-synuclein inclusions in distinct brain areas like the hippocampus, the cortex, and amygdala.[14][15] However, according to the Braak staging, α-synuclein aggregates initially develop in the olfactory bulb and the lower brainstem; propagating towards the higher brainstem and the substantia nigra; reaching the mesocortex and the thalamus; and, ultimately covering the neocortex.[8] Braak staging is a widely used method to measure the stage of pathology (stage 1 being the lowest level of pathology and stage 4 being the highest) of Parkinson's disease; used both in basic research and clinically.[13]
| Stage | Brain regions of α-synuclein pathology |
|---|---|
| 1 | olfactory bulb and lower brainstem |
| 2 | higher brainstem and substantia nigra |
| 3 | mesocortex and thalamus (at this stage motor deficits develop) |
| 4 | neocortex |
There are proposed mechanisms by which α-synuclein acts, in terms of pathology, one being the inhibition of the autophagy-lysosome pathway. This pathway is highly important as it is responsible for intracellular degradation.[13][16] Therefore, as α-synuclein fibrils inhibits the function of autophagy impairing the removal of aggregated protein, there is the production of more α-synuclein inclusions since it cannot be degraded. Other pathological mechanisms include the oxidative stress, dysfunction of the mitochondria, and neuroinflammation.[13]
Alpha-synuclein preformed fibrils
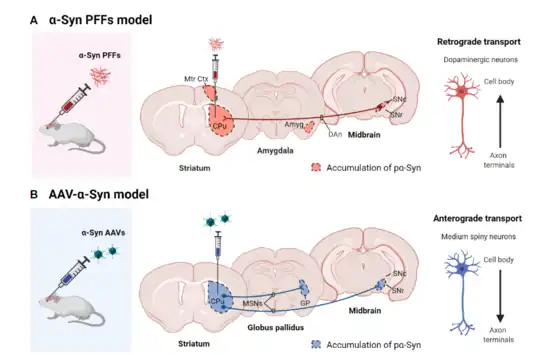
The pre-formed fibril model was developed as a way to study the propagation of α-synuclein. This model consists of injecting extracellular α-synuclein fibrils via stereotaxic injections to induce intracellular α-synuclein aggregation. Consequently, this will induce parkinsonian phenotypes.
The α-synuclein pre-formed fibrils (PFFs) are made in vitro utilizing recombinant α-synuclein monomers which will aggregate and form fibrils.[13] The fibrils can then be manipulated to form different conformations like being sonicated to form short fibrils or form heterogenous mixes of fibrils with oligomers and monomers.[15] Once the fibrils are generated, they can be injected into the brain, where hyperphosphorylation of endogenous α- synuclein (pα-syn) will occur and induce aggregation, forming cytoplasmic Lewy body and Lewy neurite inclusions. This method can be injected in brain regions like the SNpc and the cortex, however, the most common region to inject PFFs is into the striatum. Moreover, the spread of α-synuclein PFFs to brain regions occur through the uptake of the fibrils by dopamine neuron terminals that make their way up to the soma in the SNpc (Figure 9).[13] A limitation to the pre-formed fibril model is that although it is a widely used model, it lacks overt neurodegeneration.[11]
Alpha-synuclein viral vector mediated overexpression
The different synuclein models that have been widely used have also faced challenges of targeting the fibrils to the SNpc, thus, lacking abundant neurodegeneration. Through the viral vector-mediated delivery of alpha-synuclein, the vectors can target the dopaminergic neurons directly. Vectors like lentivirus and adeno-associated virus have been used in this method.[13][17] This method allows for targeting of nigrostriatal neurons, where α-synuclein protein can be overexpressed and there can be a production of alpha-synuclein- leading to accumulated phosphorylated α-synuclein in the SNpc, and overt dopaminergic neurodegeneration, including loss of dopamine terminals in the striatum.[17] Moreover, the use of the viral vectors, allows for a longer lasting expression of α-synuclein. Delivery of the α-synuclein through the viral vector is conducted through stereotaxic injections into the brain similar to the injections of α-synuclein pre-formed fibrils.[17][13] In addition, the optimal pα-synuclein expression in this method is around week 4 post-injection.[13]
In contrast to the PFF model, the α-synuclein inclusions are nuclear and demonstrates an anterograde transport in which the pα-syn travels from the soma of the neuron to the terminals, where expression are maintained within medium spiny neurons.[13]
LRRK-2
Leucine-rich repeat kinase (LRRK2) is a protein, that when mutated, is implicated in PD pathology. It is associated with both familial (most prevalent causes of familial PD) and sporadic PD. There are key mutations of the LRRK2 protein, like G2019s which is the most common missense mutation and R1441C/G.[14] Most studies have been conducted on C.elegans, Drosophila, and rodents (mice and rats). It is still unclear as to the mechanism of action of LRRK2, however, the kinase activity is of importance and its ability to function as a GTPase is also a factor in its neurotoxicity.[18] Unlike the other Parkinson's disease genetic models, LRRK2 can exhibit both Lewy body pathology and tau pathology, but it is also unclear as to its relationship.[18] On the other hand, results from LRRK2 mutation studies have demonstrated deficits in dopamine transmission, as well as axonal degeneration. Similar to other genetic animal models, LRRK2 mutations also produce dopaminergic neuron loss in the substantia nigra and Lewy body pathology. LRRK2 knockout models have also been studied and show the increase of protein aggregation and accumulation which also includes α-synuclein; but, it does not decrease degeneration of nigrostriatal neurons.[19] A limitation to the LRRK2 animal model is that although there is a loss in dopaminergic neurons, neurodegeneration is very low.
PINK1
Pten-Induced Kinase 1 (PINK1) mutations are associated with autosomal recessive parkinsonism.[4] It is a neuroprotective kinase predominantly found in the mitochondria and cytoplasmic areas of the cell. PINK1 is also a serine/threonine protein kinase and is associated with the mitochondria.[20] PINK1, in research studies, is generally used as a knockout (KO) model. The mechanism of action of this gene involves the recruitment of the Parkin gene from the cytoplasm to the mitochondria. Once recruited, this leads to augmented ubiquitin activity and therefore induces mitophagy. Mitophagy is a pathway in which the mitochondria is degraded.[21] Both PINK1 and Parkin share functions in the same pathway, therefore, their activities are similar.[19] Some studies have demonstrated expression of the PINK1 mutation in rodents, inducing dopaminergic neuron loss and motor defects.[14] Other studies are more associated with PINK1 KO. PINK1 knockouts show reduction in dopamine levels in the striatum.[4] They express very low levels of dopaminergic neuron loss and do not present the formation of Lewy bodies. However, the KO models demonstrate mitochondria dysfunction and oxidative stress. On the other hand, studies are demonstrating loss of dopaminergic neurons and showing motor deficits in rats.[19]
DJ-1
Protein Deglycase (DJ-1) mutations are associated with recessive forms of familial parkinsonism.[14] It is a molecular chaperone that undergoes reduction-oxidation (redox) reaction and plays a major role in the inhibition of alpha synuclein aggregate formation.[4] It is believed that this is possible due to DJ-1 antioxidant properties, therefore, inhibiting oxidative stress in the cell which is what induces pathological phenotypes. To demonstrate the proposed neuroprotective properties of DJ-1, knockout studies of this gene have shown motor deficits in mice, less dopamine levels in the striatum, and no evidence of Lewy body aggregation.[14] In addition to the knockout model, DJ-1 is very sensitive to neurotoxins (MPTP, 6-OHDA, etc.). Studies have demonstrated that under those conditions, DJ-1 expresses dopaminergic neuron loss in the SNpc and motor defects.[19]
Summary
Table 1[8][6][22][11] represents a summary of the PD animal models and details regarding their mechanisms of action, pathogenesis, and limitations.
| Model Type | PD association | Category | Mechanism of Action | PD Pathogenesis | Limitations |
|---|---|---|---|---|---|
| 6-OHDA | N/A | Neurotoxin | -oxidative damage -inhibition of mitochondrial respiratory chain complex I |
-Decreased striatal dopaminergic neurons -Decreased dopamine terminals -Decreased TH+ neurons in striatum and SNpc -Motor deficits |
Rapid neuronal death (apoptosis); cannot study pathological progression |
| MPTP | Neurotoxin | inhibition of mitochondrial respiratory chain complex I | -Decreased dopaminergic neurons
-Decreased striatal dopaminergic neurons -Mild decrease in motor deficits |
Rapid neuronal death (apoptosis); cannot study pathological progression | |
| Rotenone | Neurotoxin (Herbicide) | inhibition of mitochondrial respiratory chain complex I | -Decreased dopaminergic neurons
-Increased alpha synuclein -Motor deficits |
Extremely toxic - rapid neuronal death | |
| Paraquat | Neurotoxin (Herbicide) | -inhibition of mitochondrial respiratory chain complex I | -Decreased TH+ neurons in striatum
-Motor deficits |
Extremely toxic - rapid neuronal death | |
| Alpha-synuclein
(SNCA gene) |
sporadic PD | Genetic | -impairs autophagy-lysosome pathway -dysregulates mitochondrial function and aggregates in organelles -induces high oxidative stress |
-Lewy body aggregation | Low levels of dopaminergic neuron loss |
| Leucine-rich repear kinase 2 (LRRK2 gene) | familial & sporadic PD
*mostly associated with familial |
Genetic | Mechanism is unclear
-Kinase activity is proposed as well as GTPase activity |
-Lewy body aggregation
-Decreased dopaminergic neurons -Motor deficits |
Low levels of dopaminergic neuron loss |
| Pten-induced kinase 1 (PINK1 gene) | recessive familial PD | Genetic | -dysregulates mitochondrial function
-activates mitophagy with Parkin gene interaction |
-Decreased dopaminergic neurons
-Motor deficits |
Low levels of dopaminergic neuron loss |
| Protein deglycase (DJ-1 gene) | recessive familial PD | Genetic | -reduction-oxidation (RedOx) reaction
-neuroprotective properties |
DJ-1 KO:
decreased dopaminergic neurons in SNpc |
DJ-1 KO:
-Low levels of dopaminergic loss in the SNpc -Absence of Lewy body aggregation |
References
- "Parkinson's Disease". National Institute on Aging. Retrieved 2020-12-12.
- "Neurodegenerative Diseases". National Institute of Environmental Health Sciences. Retrieved 2020-12-12.
- Konnova, Elena A.; Swanberg, Maria (2018), Stoker, Thomas B.; Greenland, Julia C. (eds.), "Animal Models of Parkinson's Disease", Parkinson’s Disease: Pathogenesis and Clinical Aspects, Brisbane (AU): Codon Publications, ISBN 978-0-9944381-6-4, PMID 30702844, retrieved 2020-09-28
- Blesa, Javier; Phani, Sudarshan; Jackson-Lewis, Vernice; Przedborski, Serge (2012-03-28). "Classic and New Animal Models of Parkinson's Disease". Journal of Biomedicine and Biotechnology. 2012: 845618. doi:10.1155/2012/845618. PMC 3321500. PMID 22536024.
- Hernandez-Baltazar, Daniel; Nadella, Rasajna; Rovirosa-Hernandez, Maria de Jesus; Zavala-Flores, Laura Mireya; Jarquin, Christian de Jesus Rosas (2018-05-23), Bartholomew, Ibeh (ed.), "Animal Model of Parkinson Disease: Neuroinflammation and Apoptosis in the 6-Hydroxydopamine-Induced Model", Experimental Animal Models of Human Diseases - An Effective Therapeutic Strategy, InTech, doi:10.5772/intechopen.71271, ISBN 978-1-78923-164-9, retrieved 2020-11-16
- Prasad, E. Maruthi; Hung, Shih-Ya (2020-10-16). "Behavioral Tests in Neurotoxin-Induced Animal Models of Parkinson's Disease". Antioxidants. 9 (10): 1007. doi:10.3390/antiox9101007. ISSN 2076-3921. PMC 7602991. PMID 33081318.
- Hernandez-Baltazar, D.; Zavala-Flores, L.M.; Villanueva-Olivo, A. (2017-10-01). "The 6-hydroxydopamine model and parkinsonian pathophysiology: Novel findings in an older model". Neurología (English Edition). 32 (8): 533–539. doi:10.1016/j.nrleng.2015.06.019. ISSN 2173-5808. PMID 26304655.
- Katzung, Bertram G. (2018). Basic & Clinical Pharmacology. United States of America: McGraw-Hill Education. pp. 493–494, 1012, 1014. ISBN 978-1-259-64115-2.
- Hatcher, Jaime M.; Pennell, Kurt D.; Miller, Gary W. (June 2008). "Parkinson's disease and pesticides: a toxicological perspective". Trends in Pharmacological Sciences. 29 (6): 322–329. doi:10.1016/j.tips.2008.03.007. ISSN 0165-6147. PMC 5683846. PMID 18453001.
- Bastías-Candia, Sussy; Zolezzi, Juan M.; Inestrosa, Nibaldo C. (2019-02-01). "Revisiting the Paraquat-Induced Sporadic Parkinson's Disease-Like Model". Molecular Neurobiology. 56 (2): 1044–1055. doi:10.1007/s12035-018-1148-z. ISSN 1559-1182. PMID 29862459. S2CID 44134739.
- Ekstrand, Mats I.; Galter, Dagmar (December 2009). "The MitoPark Mouse – An animal model of Parkinson's disease with impaired respiratory chain function in dopamine neurons". Parkinsonism & Related Disorders. 15: S185–S188. doi:10.1016/S1353-8020(09)70811-9. PMID 20082987.
- Gao, Lina; Yuan, Huiya; Xu, Enyu; Liu, Junting (2020-02-04). "Toxicology of paraquat and pharmacology of the protective effect of 5-hydroxy-1-methylhydantoin on lung injury caused by paraquat based on metabolomics". Scientific Reports. 10 (1): 1790. Bibcode:2020NatSR..10.1790G. doi:10.1038/s41598-020-58599-y. ISSN 2045-2322. PMC 7000692. PMID 32019966.
- Gómez-Benito, Mónica; Granado, Noelia; García-Sanz, Patricia; Michel, Anne; Dumoulin, Mireille; Moratalla, Rosario (2020-04-23). "Modeling Parkinson's Disease With the Alpha-Synuclein Protein". Frontiers in Pharmacology. 11: 356. doi:10.3389/fphar.2020.00356. ISSN 1663-9812. PMC 7191035. PMID 32390826.
- Chia, Shyh Jenn; Tan, Eng-King; Chao, Yin-Xia (2020-04-02). "Historical Perspective: Models of Parkinson's Disease". International Journal of Molecular Sciences. 21 (7): 2464. doi:10.3390/ijms21072464. ISSN 1422-0067. PMC 7177377. PMID 32252301.
- Froula, Jessica M.; Castellana-Cruz, Marta; Anabtawi, Nadia M.; Camino, José D.; Chen, Serene W.; Thrasher, Drake R.; Freire, Jennifer; Yazdi, Allen A.; Fleming, Sheila; Dobson, Christopher M.; Kumita, Janet R. (2019-07-05). "Defining α-synuclein species responsible for Parkinson's disease phenotypes in mice". Journal of Biological Chemistry. 294 (27): 10392–10406. doi:10.1074/jbc.RA119.007743. ISSN 0021-9258. PMC 6615698. PMID 31142553.
- Yim, Willa Wen-You; Mizushima, Noboru (2020-02-11). "Lysosome biology in autophagy". Cell Discovery. 6 (1): 6. doi:10.1038/s41421-020-0141-7. ISSN 2056-5968. PMC 7010707. PMID 32047650. S2CID 211074041.
- Visanji, Naomi P.; Brotchie, Jonathan M.; Kalia, Lorraine V.; Koprich, James B.; Tandon, Anurag; Watts, Joel C.; Lang, Anthony E. (November 2016). "α-Synuclein-Based Animal Models of Parkinson's Disease: Challenges and Opportunities in a New Era". Trends in Neurosciences. 39 (11): 750–762. doi:10.1016/j.tins.2016.09.003. PMID 27776749. S2CID 205405488.
- Islam, Md. Shariful; Moore, Darren J. (2017-02-08). "Mechanisms of LRRK2-dependent neurodegeneration: role of enzymatic activity and protein aggregation". Biochemical Society Transactions. 45 (1): 163–172. doi:10.1042/BST20160264. ISSN 0300-5127. PMC 5521802. PMID 28202670.
- Jagmag, Shail A.; Tripathi, Naveen; Shukla, Sunil D.; Maiti, Sankar; Khurana, Sukant (2016-01-19). "Evaluation of Models of Parkinson's Disease". Frontiers in Neuroscience. 9: 503. doi:10.3389/fnins.2015.00503. ISSN 1662-453X. PMC 4718050. PMID 26834536.
- Kin, Kyohei; Yasuhara, Takao; Kameda, Masahiro; Date, Isao (2019-10-30). "Animal Models for Parkinson's Disease Research: Trends in the 2000s". International Journal of Molecular Sciences. 20 (21): 5402. doi:10.3390/ijms20215402. ISSN 1422-0067. PMC 6862023. PMID 31671557.
- Ding, Wen-Xing; Yin, Xiao-Ming (2012-07-01). "Mitophagy: mechanisms, pathophysiological roles, and analysis". Biological Chemistry. 393 (7): 547–564. doi:10.1515/hsz-2012-0119. ISSN 1437-4315. PMC 3630798. PMID 22944659.
- Rodriguez-Pallares, J.; Parga, J. A.; Muñoz, A.; Rey, P.; Guerra, M. J.; Labandeira-Garcia, J. L. (2007-06-15). "Mechanism of 6-hydroxydopamine neurotoxicity: the role of NADPH oxidase and microglial activation in 6-hydroxydopamine-induced degeneration of dopaminergic neurons". Journal of Neurochemistry. 103 (1): 070615193023005––. doi:10.1111/j.1471-4159.2007.04699.x. ISSN 0022-3042. PMID 17573824. S2CID 20714033.