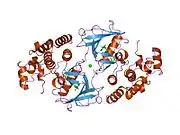
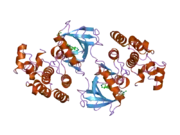
BRAF (gene)
BRAF is a human gene that encodes a protein called B-Raf. The gene is also referred to as proto-oncogene B-Raf and v-Raf murine sarcoma viral oncogene homolog B, while the protein is more formally known as serine/threonine-protein kinase B-Raf.[5][6]
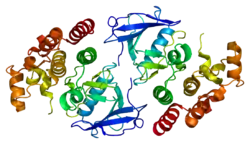




The B-Raf protein is involved in sending signals inside cells which are involved in directing cell growth. In 2002, it was shown to be mutated in some human cancers.[7]
Certain other inherited BRAF mutations cause birth defects.
Drugs that treat cancers driven by BRAF mutations have been developed. Two of these drugs, vemurafenib[8] and dabrafenib are approved by FDA for treatment of late-stage melanoma. Vemurafenib was the first approved drug to come out of fragment-based drug discovery.[9]
Function
B-Raf is a member of the Raf kinase family of growth signal transduction protein kinases. This protein plays a role in regulating the MAP kinase/ERKs signaling pathway, which affects cell division, differentiation, and secretion.[10]
Structure
B-Raf is a 766-amino acid, regulated signal transduction serine/threonine-specific protein kinase. Broadly speaking, it is composed of three conserved domains characteristic of the Raf kinase family: conserved region 1 (CR1), a Ras-GTP-binding[11] self-regulatory domain, conserved region 2 (CR2), a serine-rich hinge region, and conserved region 3 (CR3), a catalytic protein kinase domain that phosphorylates a consensus sequence on protein substrates.[12] In its active conformation, B-Raf forms dimers via hydrogen-bonding and electrostatic interactions of its kinase domains.[13]
CR1
Conserved region 1 autoinhibits B-Raf's kinase domain (CR3) so that B-Raf signaling is regulated rather than constitutive.[12] Residues 155–227[14] make up the Ras-binding domain (RBD), which binds to Ras-GTP's effector domain to release CR1 and halt kinase inhibition. Residues 234–280 comprise a phorbol ester/DAG-binding zinc finger motif that participates in B-Raf membrane docking after Ras-binding.[14][15]
CR2
Conserved Region 2 (CR2) provides a flexible linker that connects CR1 and CR3 and acts as a hinge.
CR3
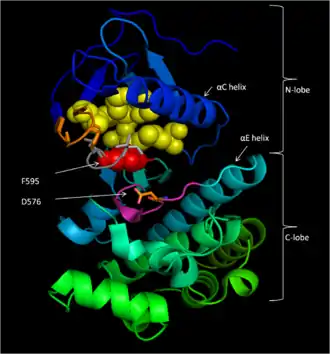
Conserved Region 3 (CR3), residues 457–717,[14] makes up B-Raf's enzymatic kinase domain. This largely conserved structure[16] is bi-lobal, connected by a short hinge region.[17] The smaller N-lobe (residues 457–530) is primarily responsible for ATP binding while the larger C-lobe (residues 535–717) binds substrate proteins.[16] The active site is the cleft between the two lobes, and the catalytic Asp576 residue is located on the C-lobe, facing the inside of this cleft.[14][16]
Subregions
P-Loop
The P-loop of B-Raf (residues 464–471) stabilizes the non-transferable phosphate groups of ATP during enzyme ATP-binding. Specifically, S467, F468, and G469 backbone amides hydrogen-bond to the β-phosphate of ATP to anchor the molecule. B-Raf functional motifs have been determined by analyzing the homology of PKA analyzed by Hanks and Hunter to the B-Raf kinase domain.[16]
Nucleotide-Binding Pocket
V471, C532, W531, T529, L514, and A481 form a hydrophobic pocket within which the adenine of ATP is anchored through Van der Waals attractions upon ATP binding.[16][18]
Catalytic Loop
Residues 574–581 compose a section of the kinase domain responsible for supporting the transfer of the γ-phosphate of ATP to B-Raf's protein substrate. In particular, D576 acts as a proton acceptor to activate the nucleophilic hydroxyl oxygen on substrate serine or threonine residues, allowing the phosphate transfer reaction to occur mediated by base-catalysis.[16]
DFG Motif
D594, F595, and G596 compose a motif central to B-Raf's function in both its inactive and active state. In the inactive state, F595 occupies the nucleotide-binding pocket, prohibiting ATP from entering and decreasing the likelihood of enzyme catalysis.[13][18][19] In the active state, D594 chelates the divalent magnesium cation that stabilizes the β- and γ-phosphate groups of ATP, orienting the γ-phosphate for transfer.[16]
Activation Loop
Residues 596–600 form strong hydrophobic interactions with the P-loop in the inactive conformation of the kinase, locking the kinase in its inactive state until the activation loop is phosphorylated, destabilizing these interactions with the presence of negative charge. This triggers the shift to the active state of the kinase. Specifically, L597 and V600 of the activation loop interact with G466, F468, and V471 of the P-loop to keep the kinase domain inactive until it is phosphorylated.[17]
Enzymology
B-Raf is a serine/threonine-specific protein kinase. As such, it catalyzes the phosphorylation of serine and threonine residues in a consensus sequence on target proteins by ATP, yielding ADP and a phosphorylated protein as products.[16] Since it is a highly regulated signal transduction kinase, B-Raf must first bind Ras-GTP before becoming active as an enzyme.[15] Once B-Raf is activated, a conserved protein kinase catalytic core phosphorylates protein substrates by promoting the nucleophilic attack of the activated substrate serine or threonine hydroxyl oxygen atom on the γ-phosphate group of ATP through bimolecular nucleophilic substitution.[16][20][21][22]
Relieving CR1 autoinhibition
The kinase (CR3) domain of human Raf kinases is inhibited by two mechanisms: autoinhibition by its own regulatory Ras-GTP-binding CR1 domain and a lack of post-translational phosphorylation of key serine and tyrosine residues (S338 and Y341 for c-Raf) in the CR2 hinge region. During B-Raf activation, the protein's autoinhibitory CR1 domain first binds Ras-GTP's effector domain to the CR1 Ras-binding domain (RBD) to release the kinase CR3 domain like other members of the human Raf kinase family. The CR1-Ras interaction is later strengthened through the binding of the cysteine-rich subdomain (CRD) of CR1 to Ras and membrane phospholipids.[12] Unlike A-Raf and C-Raf, which must be phosphorylated on hydroxyl-containing CR2 residues before fully releasing CR1 to become active, B-Raf is constituitively phosphorylated on CR2 S445.[23] This allows the negatively charged phosphoserine to immediately repel CR1 through steric and electrostatic interactions once the regulatory domain is unbound, freeing the CR3 kinase domain to interact with substrate proteins.
CR3 domain activation
After the autoinhibitory CR1 regulatory domain is released, B-Raf's CR3 kinase domain must change to its ATP-binding active conformer before it can catalyze protein phosphorylation. In the inactive conformation, F595 of the DFG motif blocks the hydrophobic adenine binding pocket while activation loop residues form hydrophobic interactions with the P-loop, stopping ATP from accessing its binding site. When the activation loop is phosphorylated, the negative charge of the phosphate is unstable in the hydrophobic environment of the P-loop. As a result, the activation loop changes conformation, stretching out across the C-lobe of the kinase domain. In this process, it forms stabilizing β-sheet interactions with the β6 strand. Meanwhile, the phosphorylated residue approaches K507, forming a stabilizing salt bridge to lock the activation loop into place. The DFG motif changes conformation with the activation loop, causing F595 to move out of the adenine nucleotide binding site and into a hydrophobic pocket bordered by the αC and αE helices. Together, DFG and activation loop movement upon phosphorylation open the ATP binding site. Since all other substrate-binding and catalytic domains are already in place, phosphorylation of the activation loop alone activates B-Raf's kinase domain through a chain reaction that essentially removes a lid from an otherwise-prepared active site.[17]
Mechanism of catalysis
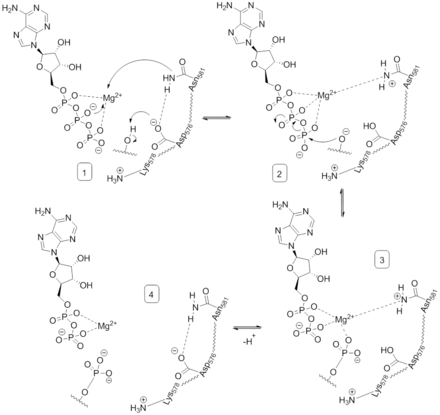
To effectively catalyze protein phosphorylation via the bimolecular substitution of serine and threonine residues with ADP as a leaving group, B-Raf must first bind ATP and then stabilize the transition state as the γ-phosphate of ATP is transferred.[16]
ATP binding
B-Raf binds ATP by anchoring the adenine nucleotide in a nonpolar pocket (yellow, Figure 1) and orienting the molecule through hydrogen-bonding and electrostatic interactions with phosphate groups. In addition to the P-loop and DFG motif phosphate binding described above, K483 and E501 play key roles in stabilizing non-transferable phosphate groups. The positive charge on the primary amine of K483 allows it to stabilize the negative charge on ATP α- and β-phosphate groups when ATP binds. When ATP is not present, the negative charge of the E501 carboxyl group balances this charge.[16][17]
Phosphorylation
Once ATP is bound to the B-Raf kinase domain, D576 of the catalytic loop activates a substrate hydroxyl group, increasing its nucleophilicity to kinetically drive the phosphorylation reaction while other catalytic loop residues stabilize the transition state.(Figure 2). N581 chelates the divalent magnesium cation associated with ATP to help orient the molecule for optimal substitution. K578 neutralizes the negative charge on the γ-phosphate group of ATP so that the activated ser/thr substrate residue won't experience as much electron-electron repulsion when attacking the phosphate. After the phosphate group is transferred, ADP and the new phosphoprotein are released.[16]
Inhibitors
Since constitutively active B-Raf mutants commonly cause cancer (see Clinical Significance) by excessively signaling cells to grow, inhibitors of B-Raf have been developed for both the inactive and active conformations of the kinase domain as cancer therapeutic candidates.[17][18][19]
Sorafenib
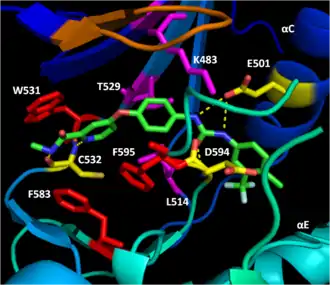
BAY43-9006 (Sorafenib, Nexavar) is a V600E mutant B-Raf and C-Raf inhibitor approved by the FDA for the treatment of primary liver and kidney cancer. Bay43-9006 disables the B-Raf kinase domain by locking the enzyme in its inactive form. The inhibitor accomplishes this by blocking the ATP binding pocket through high-affinity for the kinase domain. It then binds key activation loop and DFG motif residues to stop the movement of the activation loop and DFG motif to the active conformation. Finally, a trifluoromethyl phenyl moiety sterically blocks the DFG motif and activation loop active conformation site, making it impossible for the kinase domain to shift conformation to become active.[17]
The distal pyridyl ring of BAY43-9006 anchors in the hydrophobic nucleotide-binding pocket of the kinase N-lobe, interacting with W531, F583, and F595. The hydrophobic interactions with catalytic loop F583 and DFG motif F595 stabilize the inactive conformation of these structures, decreasing the likelihood of enzyme activation. Further hydrophobic interaction of K483, L514, and T529 with the center phenyl ring increase the affinity of the kinase domain for the inhibitor. Hydrophobic interaction of F595 with the center ring as well decreases the energetic favorability of a DFG conformation switch further. Finally, polar interactions of BAY43-9006 with the kinase domain continue this trend of increasing enzyme affinity for the inhibitor and stabilizing DFG residues in the inactive conformation. E501 and C532 hydrogen bond the urea and pyridyl groups of the inhibitor respectively while the urea carbonyl accepts a hydrogen bond from D594's backbone amide nitrogen to lock the DFG motif in place.[17]
The trifluoromethyl phenyl moiety cements the thermodynamic favorability of the inactive conformation when the kinase domain is bound to BAY43-9006 by sterically blocking the hydrophobic pocket between the αC and αE helices that the DFG motif and activation loop would inhabit upon shifting to their locations in the active conformation of the protein.[17]
Vemurafenib
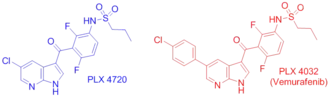
PLX4032 (Vemurafenib) is a V600 mutant B-Raf inhibitor approved by the FDA for the treatment of late-stage melanoma.[13] Unlike BAY43-9006, which inhibits the inactive form of the kinase domain, Vemurafenib inhibits the active "DFG-in" form of the kinase,[18][19] firmly anchoring itself in the ATP-binding site. By inhibiting only the active form of the kinase, Vemurafenib selectively inhibits the proliferation of cells with unregulated B-Raf, normally those that cause cancer.
Since Vemurafenib only differs from its precursor, PLX4720, in a phenyl ring added for pharmacokinetic reasons,[19] PLX4720's mode of action is equivalent to Vemurafenib's. PLX4720 has good affinity for the ATP binding site partially because its anchor region, a 7-azaindole bicyclic, only differs from the natural adenine that occupies the site in two places where nitrogen atoms have been replaced by carbon. This enables strong intermolecular interactions like N7 hydrogen bonding to C532 and N1 hydrogen bonding to Q530 to be preserved. Excellent fit within the ATP-binding hydrophobic pocket (C532, W531, T529, L514, A481) increases binding affinity as well. Ketone linker hydrogen bonding to water and difluoro-phenyl fit in a second hydrophobic pocket (A481, V482, K483, V471, I527, T529, L514, and F583) contribute to the exceptionally high binding affinity overall. Selective binding to active Raf is accomplished by the terminal propyl group that binds to a Raf-selective pocket created by a shift of the αC helix. Selectivity for the active conformation of the kinase is further increased by a pH-sensitive deprotonated sulfonamide group that is stabilized by hydrogen bonding with the backbone peptide NH of D594 in the active state. In the inactive state, the inhibitor's sulfonamide group interacts with the backbone carbonyl of that residue instead, creating repulsion. Thus, Vemurafenib binds preferentially to the active state of B-Raf's kinase domain.[18][19]
Clinical significance
Mutations in the BRAF gene can cause disease in two ways. First, mutations can be inherited and cause birth defects. Second, mutations can appear later in life and cause cancer, as an oncogene.
Inherited mutations in this gene cause cardiofaciocutaneous syndrome, a disease characterized by heart defects, mental retardation and a distinctive facial appearance.[24]
Mutations in this gene have been found in cancers, including non-Hodgkin lymphoma, colorectal cancer, malignant melanoma, papillary thyroid carcinoma, non-small-cell lung carcinoma, adenocarcinoma of the lung, brain tumors including glioblastoma and pleomorphic xanthoastrocytoma as well as inflammatory diseases like Erdheim-Chester disease.[10]
The V600E mutation of the BRAF gene has been associated with hairy cell leukemia in numerous studies and has been suggested for use in screening for Lynch syndrome to reduce the number of patients undergoing unnecessary MLH1 sequencing.[25][26]
Mutants
More than 30 mutations of the BRAF gene associated with human cancers have been identified. The frequency of BRAF mutations varies widely in human cancers, from more than 80% in melanomas and nevi, to as little as 0–18% in other tumors, such as 1–3% in lung cancers and 5% in colorectal cancer.[27] In 90% of the cases, thymine is substituted with adenine at nucleotide 1799. This leads to valine (V) being substituted for by glutamate (E) at codon 600 (now referred to as V600E) in the activation segment that has been found in human cancers.[28] This mutation has been widely observed in papillary thyroid carcinoma, colorectal cancer, melanoma and non-small-cell lung cancer.[29][30][31][32][33][34][35] BRAF-V600E mutation are present in 57% of Langerhans cell histiocytosis patients.[36] The V600E mutation is a likely driver mutation in 100% of cases of hairy cell leukaemia.[37] High frequency of BRAF V600E mutations have been detected in ameloblastoma, a benign but locally infiltrative odontogenic neoplasm.[38] The V600E mutation may also be linked, as a single-driver mutation (a genetic 'smoking gun') to certain cases of papillary craniopharyngioma development.[39]
Other mutations which have been found are R461I, I462S, G463E, G463V, G465A, G465E, G465V, G468A, G468E, G469R, N580S, E585K, D593V, F594L, G595R, L596V, T598I, V599D, V599E, V599K, V599R, V600K, A727V, etc. and most of these mutations are clustered to two regions: the glycine-rich P loop of the N lobe and the activation segment and flanking regions.[17] These mutations change the activation segment from inactive state to active state, for example in the previous cited paper it has been reported that the aliphatic side chain of Val599 interacts with the phenyl ring of Phe467 in the P loop. Replacing the medium-sized hydrophobic Val side chain with a larger and charged residue as found in human cancer(Glu, Asp, Lys, or Arg) would be expected to destabilize the interactions that maintain the DFG motif in an inactive conformation, so flipping the activation segment into the active position. Depending on the type of mutation the kinase activity towards MEK may also vary. Most of the mutants stimulate enhanced B-Raf kinase activity toward MEK. However, a few mutants act through a different mechanism because although their activity toward MEK is reduced, they adopt a conformation that activates wild-type C-RAF, which then signals to ERK.
BRAF-V600E
- BRAFV600E mutation confers a poor prognosis in metastatic colorectal cancer. Upon targeted inhibition of BRAF and/or EGFR in BRAF V600E colorectal cancer, SRC kinases become activated in a systematic manner. Remarkably, concurrent targeting of SRC alongside BRAF and EGFR has demonstrated increased treatment effectiveness. The compensatory activation of SRC kinases is mediated by an autocrine prostaglandin E2 loop, which can be blocked using cyclooxygenase-2 (COX2) inhibitors. Notably, the simultaneous targeting of COX2 with BRAF and EGFR has shown significant potential in preclinical models, leading to a sustained suppression of tumor growth.[40]
- BRAF V600E is a determinant of sensitivity to proteasome inhibitors. Vulnerability to proteasome inhibitors is dependent on persistent BRAF signaling, because BRAF-V600E blockade by PLX4720 reversed sensitivity to carfilzomib in BRAF-mutant colorectal cancer cells. Proteasome inhibition might represent a valuable targeting strategy in BRAF V600E-mutant colorectal tumors.[41]
BRAF inhibitors
As mentioned above, some pharmaceutical firms are developing specific inhibitors of mutated B-raf protein for anticancer use because BRAF is a well-understood, high yield target.[18][42] Vemurafenib (RG7204 or PLX4032) was licensed by the US Food and Drug Administration as Zelboraf for the treatment of metastatic melanoma in August 2011 based on Phase III clinical data. Improved survival was seen, as well as a response rate to treatment of 53%, compared to from 7-12% with the former best chemotherapeutic treatment, dacarbazine.[43] In clinical trials, B-Raf increased metastatic melanoma patient chance of survival. In spite of the drug's high efficacy, 20% of tumors still develop resistance to the treatment. In mice, 20% of tumors become resistant after 56 days.[44] While the mechanisms of this resistance are still disputed, some hypotheses include the overexpression of B-Raf to compensate for high concentrations of Vemurafenib[44] and upstream upregulation of growth signaling.[45]
More general B-Raf inhibitors include GDC-0879, PLX-4720, Sorafenib, dabrafenib and encorafenib.
panRAF inhibitors
Belvarafenib is classified as a panRAF inhibitor. A panRAF inhibitor blocks the catalytic function of both proteins in the dimer.[46]
Interactions
BRAF (gene) has been shown to interact with:
References
- GRCh38: Ensembl release 89: ENSG00000157764 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000002413 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Sithanandam G, Kolch W, Duh FM, Rapp UR (December 1990). "Complete coding sequence of a human B-raf cDNA and detection of B-raf protein kinase with isozyme specific antibodies". Oncogene. 5 (12): 1775–1780. PMID 2284096.
- Sithanandam G, Druck T, Cannizzaro LA, Leuzzi G, Huebner K, Rapp UR (April 1992). "B-raf and a B-raf pseudogene are located on 7q in man". Oncogene. 7 (4): 795–799. PMID 1565476.
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. (June 2002). "Mutations of the BRAF gene in human cancer". Nature. 417 (6892): 949–954. Bibcode:2002Natur.417..949D. doi:10.1038/nature00766. PMID 12068308. S2CID 3071547.
- "FDA Approves Zelboraf (Vemurafenib) and Companion Diagnostic for BRAF Mutation-Positive Metastatic Melanoma, a Deadly Form of Skin Cancer" (Press release). Genentech. Retrieved 2011-08-17.
- Erlanson DA, Fesik SW, Hubbard RE, Jahnke W, Jhoti H (September 2016). "Twenty years on: the impact of fragments on drug discovery". Nature Reviews. Drug Discovery. 15 (9): 605–619. doi:10.1038/nrd.2016.109. PMID 27417849. S2CID 19634793.
- "Entrez Gene: BRAF".
- Daum G, Eisenmann-Tappe I, Fries HW, Troppmair J, Rapp UR (November 1994). "The ins and outs of Raf kinases". Trends in Biochemical Sciences. 19 (11): 474–480. doi:10.1016/0968-0004(94)90133-3. PMID 7855890.
- Cutler RE, Stephens RM, Saracino MR, Morrison DK (August 1998). "Autoregulation of the Raf-1 serine/threonine kinase". Proceedings of the National Academy of Sciences of the United States of America. 95 (16): 9214–9219. Bibcode:1998PNAS...95.9214C. doi:10.1073/pnas.95.16.9214. PMC 21318. PMID 9689060.
- Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, Hirth P (November 2012). "Vemurafenib: the first drug approved for BRAF-mutant cancer". Nature Reviews. Drug Discovery. 11 (11): 873–886. doi:10.1038/nrd3847. PMID 23060265. S2CID 9337155.
- "Serine/threonine protein kinase B-rAF". Retrieved 4 Mar 2013.
- Morrison DK, Cutler RE (April 1997). "The complexity of Raf-1 regulation". Current Opinion in Cell Biology. 9 (2): 174–179. doi:10.1016/S0955-0674(97)80060-9. PMID 9069260.
- Hanks SK, Hunter T (May 1995). "Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification". FASEB Journal. 9 (8): 576–596. doi:10.1096/fasebj.9.8.7768349. PMID 7768349. S2CID 21377422.
- Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. (March 2004). Cancer Genome Project. "Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF". Cell. 116 (6): 855–867. doi:10.1016/S0092-8674(04)00215-6. PMID 15035987. S2CID 126161.
- Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al. (February 2008). "Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity". Proceedings of the National Academy of Sciences of the United States of America. 105 (8): 3041–3046. Bibcode:2008PNAS..105.3041T. doi:10.1073/pnas.0711741105. PMC 2268581. PMID 18287029.
- Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. (September 2010). "Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma". Nature. 467 (7315): 596–599. Bibcode:2010Natur.467..596B. doi:10.1038/nature09454. PMC 2948082. PMID 20823850.
- Hanks SK, Quinn AM, Hunter T (July 1988). "The protein kinase family: conserved features and deduced phylogeny of the catalytic domains". Science. 241 (4861): 42–52. Bibcode:1988Sci...241...42H. doi:10.1126/science.3291115. PMID 3291115.
- Hanks SK (June 1991). "Eukaryotic protein kinases". Curr. Opin. Struct. Biol. 1 (3): 369–383. doi:10.1016/0959-440X(91)90035-R.
- Hanks SK, Quinn AM (1991). "[2] Protein kinase catalytic domain sequence database: Identification of conserved features of primary structure and classification of family members". Protein kinase catalytic domain sequence database: identification of conserved features of primary structure and classification of family members. Methods in Enzymology. Vol. 200. pp. 38–62. doi:10.1016/0076-6879(91)00126-H. ISBN 9780121821012. PMID 1956325.
- Mason CS, Springer CJ, Cooper RG, Superti-Furga G, Marshall CJ, Marais R (April 1999). "Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation". The EMBO Journal. 18 (8): 2137–2148. doi:10.1093/emboj/18.8.2137. PMC 1171298. PMID 10205168.
- Roberts A, Allanson J, Jadico SK, Kavamura MI, Noonan J, Opitz JM, et al. (November 2006). "The cardiofaciocutaneous syndrome". Journal of Medical Genetics. 43 (11): 833–842. doi:10.1136/jmg.2006.042796. PMC 2563180. PMID 16825433.
- Ewalt M, Nandula S, Phillips A, Alobeid B, Murty VV, Mansukhani MM, Bhagat G (December 2012). "Real-time PCR-based analysis of BRAF V600E mutation in low and intermediate grade lymphomas confirms frequent occurrence in hairy cell leukaemia". Hematological Oncology. 30 (4): 190–193. doi:10.1002/hon.1023. PMID 22246856. S2CID 204843221.
- Palomaki GE, McClain MR, Melillo S, Hampel HL, Thibodeau SN (January 2009). "EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome". Genetics in Medicine. 11 (1): 42–65. doi:10.1097/GIM.0b013e31818fa2db. PMC 2743613. PMID 19125127.
- Namba H, Nakashima M, Hayashi T, Hayashida N, Maeda S, Rogounovitch TI, et al. (September 2003). "Clinical implication of hot spot BRAF mutation, V599E, in papillary thyroid cancers". The Journal of Clinical Endocrinology and Metabolism. 88 (9): 4393–4397. doi:10.1210/jc.2003-030305. PMID 12970315.
- Tan YH, Liu Y, Eu KW, Ang PW, Li WQ, Salto-Tellez M, et al. (April 2008). "Detection of BRAF V600E mutation by pyrosequencing". Pathology. 40 (3): 295–298. doi:10.1080/00313020801911512. PMID 18428050. S2CID 32051681.
- Li WQ, Kawakami K, Ruszkiewicz A, Bennett G, Moore J, Iacopetta B (January 2006). "BRAF mutations are associated with distinctive clinical, pathological and molecular features of colorectal cancer independently of microsatellite instability status". Molecular Cancer. 5 (1): 2. doi:10.1186/1476-4598-5-2. PMC 1360090. PMID 16403224.
- Benlloch S, Payá A, Alenda C, Bessa X, Andreu M, Jover R, et al. (November 2006). "Detection of BRAF V600E mutation in colorectal cancer: comparison of automatic sequencing and real-time chemistry methodology". The Journal of Molecular Diagnostics. 8 (5): 540–543. doi:10.2353/jmoldx.2006.060070. PMC 1876165. PMID 17065421.
- Deng G, Bell I, Crawley S, Gum J, Terdiman JP, Allen BA, et al. (January 2004). "BRAF mutation is frequently present in sporadic colorectal cancer with methylated hMLH1, but not in hereditary nonpolyposis colorectal cancer". Clinical Cancer Research. 10 (1 Pt 1): 191–195. doi:10.1158/1078-0432.CCR-1118-3. PMID 14734469.
- Gear H, Williams H, Kemp EG, Roberts F (August 2004). "BRAF mutations in conjunctival melanoma". Investigative Ophthalmology & Visual Science. 45 (8): 2484–2488. doi:10.1167/iovs.04-0093. PMID 15277467.
- Maldonado JL, Fridlyand J, Patel H, Jain AN, Busam K, Kageshita T, et al. (December 2003). "Determinants of BRAF mutations in primary melanomas". Journal of the National Cancer Institute. 95 (24): 1878–1890. doi:10.1093/jnci/djg123. PMID 14679157.
- Puxeddu E, Moretti S, Elisei R, Romei C, Pascucci R, Martinelli M, et al. (May 2004). "BRAF(V599E) mutation is the leading genetic event in adult sporadic papillary thyroid carcinomas". The Journal of Clinical Endocrinology and Metabolism. 89 (5): 2414–2420. doi:10.1210/jc.2003-031425. PMID 15126572.
- Elisei R, Ugolini C, Viola D, Lupi C, Biagini A, Giannini R, et al. (October 2008). "BRAF(V600E) mutation and outcome of patients with papillary thyroid carcinoma: a 15-year median follow-up study". The Journal of Clinical Endocrinology and Metabolism. 93 (10): 3943–3949. doi:10.1210/jc.2008-0607. PMID 18682506.
- Badalian-Very G, Vergilio JA, Degar BA, Rodriguez-Galindo C, Rollins BJ (January 2012). "Recent advances in the understanding of Langerhans cell histiocytosis". British Journal of Haematology. 156 (2): 163–172. doi:10.1111/j.1365-2141.2011.08915.x. PMID 22017623. S2CID 34922416.
- Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, et al. (June 2011). "BRAF mutations in hairy-cell leukemia". The New England Journal of Medicine. 364 (24): 2305–2315. doi:10.1056/NEJMoa1014209. PMC 3689585. PMID 21663470. *Lay summary in: "Research on hairy-cell leukaemia shows the promise of new DNA-scanning technologies". Cancer Research UK. June 11, 2011.
- Kurppa KJ, Catón J, Morgan PR, Ristimäki A, Ruhin B, Kellokoski J, et al. (April 2014). "High frequency of BRAF V600E mutations in ameloblastoma". The Journal of Pathology. 232 (5): 492–498. doi:10.1002/path.4317. PMC 4255689. PMID 24374844.
- Brastianos PK, Taylor-Weiner A, Manley PE, Jones RT, Dias-Santagata D, Thorner AR, et al. (February 2014). "Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas". Nature Genetics. 46 (2): 161–165. doi:10.1038/ng.2868. PMC 3982316. PMID 24413733.*Lay summary in: Leah Eisenstadt (30 January 2014). "Single driver mutation found in rare brain tumor". BROAD Institute.
- Ruiz-Saenz A, Atreya CE, Wang C, Pan B, Dreyer CA, Brunen D, et al. (February 2023). "A reversible SRC-relayed COX2 inflammatory program drives resistance to BRAF and EGFR inhibition in BRAFV600E colorectal tumors". Nature Cancer. 4 (2): 240–256. doi:10.1038/s43018-022-00508-5. PMC 9970872. PMID 36759733.
- Zecchin D, Boscaro V, Medico E, Barault L, Martini M, Arena S, et al. (December 2013). "BRAF V600E is a determinant of sensitivity to proteasome inhibitors". Molecular Cancer Therapeutics. 12 (12): 2950–2961. doi:10.1158/1535-7163.MCT-13-0243. hdl:2318/140935. PMID 24107445. S2CID 17012966.
- King AJ, Patrick DR, Batorsky RS, Ho ML, Do HT, Zhang SY, et al. (December 2006). "Demonstration of a genetic therapeutic index for tumors expressing oncogenic BRAF by the kinase inhibitor SB-590885". Cancer Research. 66 (23): 11100–11105. doi:10.1158/0008-5472.CAN-06-2554. PMID 17145850.
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. (June 2011). BRIM-3 Study Group. "Improved survival with vemurafenib in melanoma with BRAF V600E mutation". The New England Journal of Medicine. 364 (26): 2507–2516. doi:10.1056/NEJMoa1103782. PMC 3549296. PMID 21639808.
- Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, et al. (February 2013). "Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance". Nature. 494 (7436): 251–255. Bibcode:2013Natur.494..251D. doi:10.1038/nature11814. PMC 3930354. PMID 23302800.
- Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. (December 2010). "Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation". Nature. 468 (7326): 973–977. Bibcode:2010Natur.468..973N. doi:10.1038/nature09626. PMC 3143360. PMID 21107323.
- Degirmenci U, Yap J, Sim YR, Qin S, Hu J (2021). "Drug resistance in targeted cancer therapies with RAF inhibitors". Cancer Drug Resistance. 4 (3): 665–683. doi:10.20517/cdr.2021.36. PMID 35582307.
- Guan KL, Figueroa C, Brtva TR, Zhu T, Taylor J, Barber TD, Vojtek AB (September 2000). "Negative regulation of the serine/threonine kinase B-Raf by Akt". The Journal of Biological Chemistry. 275 (35): 27354–27359. doi:10.1074/jbc.M004371200. PMID 10869359.
- Weber CK, Slupsky JR, Kalmes HA, Rapp UR (May 2001). "Active Ras induces heterodimerization of cRaf and BRaf". Cancer Research. 61 (9): 3595–3598. PMID 11325826.
- Stang S, Bottorff D, Stone JC (June 1997). "Interaction of activated Ras with Raf-1 alone may be sufficient for transformation of rat2 cells". Molecular and Cellular Biology. 17 (6): 3047–3055. doi:10.1128/MCB.17.6.3047. PMC 232157. PMID 9154803.
- Reuter CW, Catling AD, Jelinek T, Weber MJ (March 1995). "Biochemical analysis of MEK activation in NIH3T3 fibroblasts. Identification of B-Raf and other activators". The Journal of Biological Chemistry. 270 (13): 7644–7655. doi:10.1074/jbc.270.13.7644. PMID 7706312.
- Ewing RM, Chu P, Elisma F, Li H, Taylor P, Climie S, et al. (2007). "Large-scale mapping of human protein-protein interactions by mass spectrometry". Molecular Systems Biology. 3 (1): 89. doi:10.1038/msb4100134. PMC 1847948. PMID 17353931.
- Qiu W, Zhuang S, von Lintig FC, Boss GR, Pilz RB (October 2000). "Cell type-specific regulation of B-Raf kinase by cAMP and 14-3-3 proteins". The Journal of Biological Chemistry. 275 (41): 31921–31929. doi:10.1074/jbc.M003327200. PMID 10931830.
Further reading
- Garnett MJ, Marais R (October 2004). "Guilty as charged: B-RAF is a human oncogene". Cancer Cell. 6 (4): 313–319. doi:10.1016/j.ccr.2004.09.022. PMID 15488754.
- Quiros RM, Ding HG, Gattuso P, Prinz RA, Xu X (June 2005). "Evidence that one subset of anaplastic thyroid carcinomas are derived from papillary carcinomas due to BRAF and p53 mutations". Cancer. 103 (11): 2261–2268. doi:10.1002/cncr.21073. PMID 15880523. S2CID 29665029.
- Karbowniczek M, Henske EP (November 2005). "The role of tuberin in cellular differentiation: are B-Raf and MAPK involved?". Annals of the New York Academy of Sciences. 1059 (1): 168–173. Bibcode:2005NYASA1059..168K. doi:10.1196/annals.1339.045. PMID 16382052. S2CID 39146204.
- Ciampi R, Nikiforov YE (March 2007). "RET/PTC rearrangements and BRAF mutations in thyroid tumorigenesis". Endocrinology. 148 (3): 936–941. doi:10.1210/en.2006-0921. PMID 16946010.
- Espinosa AV, Porchia L, Ringel MD (January 2007). "Targeting BRAF in thyroid cancer". British Journal of Cancer. 96 (1): 16–20. doi:10.1038/sj.bjc.6603520. PMC 2360215. PMID 17179987.
- Allanson JE, Roberts AE (8 August 2019). "Noonan Syndrome". In Pagon RA, Bird TD, Dolan CR, et al. (eds.). GeneReviews [Internet]. Seattle WA: University of Washington, Seattle. PMID 20301303.
- Rauen KA (3 March 2016) [18 January 2007]. "Cardiofaciocutaneous Syndrome". In Pagon RA, Bird TD, Dolan CR (eds.). GeneReviews [Internet]. Seattle WA: University of Washington, Seattle. PMID 20301365.
- Gelb BD, Tartaglia M (14 May 2015) [30 November 2007]. "LEOPARD Syndrome". In Pagon RA, Bird TD, Dolan CR (eds.). GeneReviews [Internet]. Seattle WA: University of Washington, Seattle. PMID 20301557.
External links
- "BRAF gene". NCI Dictionary of Cancer Terms. Retrieved 2007-11-25.
- Finding faults in BRAF — Cancer Research UK blog post about the discovery of cancer-causing BRAF mutations (incl video)
- Human BRAF genome location and BRAF gene details page in the UCSC Genome Browser.
![]() This article incorporates public domain material from Dictionary of Cancer Terms. U.S. National Cancer Institute.
This article incorporates text from the United States National Library of Medicine, which is in the public domain.
This article incorporates public domain material from Dictionary of Cancer Terms. U.S. National Cancer Institute.
This article incorporates text from the United States National Library of Medicine, which is in the public domain.
PDB gallery | |
|---|---|
|