Chordoma
Chordoma is a rare slow-growing neoplasm thought to arise from cellular remnants of the notochord. The evidence for this is the location of the tumors (along the neuraxis), the similar immunohistochemical staining patterns, and the demonstration that notochordal cells are preferentially left behind in the clivus and sacrococcygeal regions when the remainder of the notochord regresses during fetal life.
| Chordoma | |
|---|---|
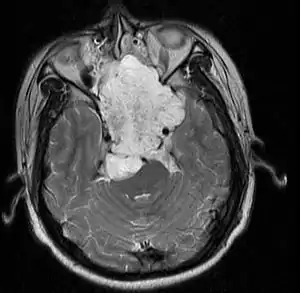 | |
| MRI of extensive clival chordoma in 17-year-old male patient, axial view. Tumor in the nasopharynx extending from nasal cavity to brainstem posteriorly is clearly visible. | |
| Specialty | Oncology |
In layman's terms, chordoma is a type of spinal cancer.[1]
Presentation
.jpg.webp)
Chordomas can arise from bone in the skull base and anywhere along the spine. The two most common locations are cranially at the clivus and in the sacrum at the bottom of the spine.[2]
Sacral chordoma is presented with chronic low back pain.[3]
Genetics
A small number of families have been reported in which multiple relatives have been affected by chordoma. In four of these families, duplication of the brachyury gene was found to be responsible for causing chordoma.[4]
A possible association with tuberous sclerosis complex (TSC1 or TSC2) has been suggested.[5]
Mechanism
- mTOR signaling is hyperactive in sporadic sacral chordomas: in one study 10 out of 10 sacral chordomas exhibited phosphorylation of Ribosomal protein s6 and EIF4EBP1 by immunohistochemistry[6]
- Partial or complete PTEN (gene) deficiency is observed in nearly all sacral chordomas[6]
- In a study of 49 chordomas Akt, TSC2, and EIF4EBP1 were phosphorylated in 92%, 96% and 98% of cases, respectively.[7]
- In a tissue microarray containing 21 chordomas Platelet-derived growth factor receptor-beta (PDGFR-b), epidermal growth factor receptor (EGFR), KIT (CD117) and HER2 were detected in 100%, 67%, 33% and 0% of cases, respectively.[8]
- The CDKN2A (p16) and CDKN2B (p15) loci on chromosome 9p21 are frequently deleted in chordomas[9] Another study found CDKN2A immunoreactivity in just 4% of cases.[7]
- 62% of chordomas express the High Molecular Weight Melanoma Associated Antigen, also known as Chondroitin sulfate proteoglycan 4 (CSPG4) which has been the target of immune therapy.[10]
- In 2009, scientists discovered that an inherited gene duplication is responsible for the familial form of this disorder.[11] Familial chordoma are rare, with an estimated rate of 0.4% in all Chordomas.[12]
Diagnosis
In 2015 the first consensus guidelines for the diagnosis and treatment of chordoma were published in The Lancet Oncology.[13] These tumors express brachyury and cytokeratin, which can be detected by immunohistochemistry.
Classification
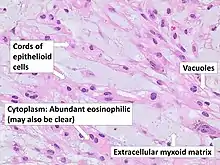
There are three histological variants of chordoma: conventional,[14] chondroid and dedifferentiated.
- The histological appearance of classical chordoma is of a lobulated tumor composed of groups of cells separated by fibrous septa. The cells have small round nuclei and abundant vacuolated cytoplasm, sometimes described as physaliferous (having bubbles or vacuoles).
- Chondroid chordomas histologically show features of both chordoma and chondrosarcoma.
Treatment
In most cases, complete surgical resection followed by radiation therapy offers the best chance of long-term control.[15] Incomplete resection of the primary tumor makes controlling the disease more difficult and increases the odds of recurrence. The decision whether complete or incomplete surgery should be performed primarily depends on the anatomical location of the tumor and its proximity to vital parts of the central nervous system.
Chordomas are relatively radioresistant, requiring high doses of radiation to be controlled. The proximity of chordomas to vital neurological structures such as the brain stem and nerves limits the dose of radiation that can safely be delivered. Therefore, highly focused radiation such as proton therapy and carbon ion therapy are more effective than conventional x-ray radiation.[16]
There are no drugs currently approved to treat chordoma, however a clinical trial conducted in Italy using a tyrosine kinase inhibitor imatinib demonstrated a modest response in some chordoma patients.[17] The same group in Italy found that the combination of imatinib and sirolimus caused a response in several patients whose tumors progressed on imatinib alone. Erlotinib-like EGFR inhibitors have been also reported to be effective in chordoma.[18] Although EGFR mutation is not present in chordoma, EGFR expression might predict response to erlotinib (as shown in report by Dr Sameer Rastogi).[18]
Prognosis
In one study, the 10-year tumor free survival rate for sacral chordoma was 46%.[20] Chondroid chordomas appear to have a more indolent clinical course.
Epidemiology
In the United States, the annual incidence of chordoma is approximately 1 in one million (300 new patients each year).[21]
Sacral chordomas make up 2 to 4% of all primary bone tumours and 44% of all primary sacral tumours, thus making it the most common malignant sacral tumour. About 50 to 60% of chordomas are located in the sacrococcygeal region. Males aged between 40 and 50 years are twice as more common than women to get sacral chordoma.[3]
There are currently no known environmental risk factors for chordoma. As noted above germline duplication of brachyury has been identified as a major susceptibility mechanism in several chordoma families.[22]
While most people with chordoma have no other family members with the disease, rare occurrences of multiple cases within families have been documented. This suggests that some people may be genetically predisposed to develop chordoma. Because genetic or hereditary risk factors for chordoma may exist, scientists at the National Cancer Institute are conducting a Familial Chordoma Study to search for genes involved in the development of this tumor.[23]
Society
Expert Recommendations for the Diagnosis and Treatment of Chordoma is a handbook produced by the Chordoma Foundation, which summarizes recommendations developed by a group of over 40 leading doctors who specialize in caring for chordoma patients. It is available electronically in English, Chinese, Italian, Dutch, and Spanish and hardcopies are available in English and Spanish.[24]
Notable cases
NFL player Craig Heyward was treated for a chordoma in 1998, which ended his career. While initially thought to be successfully removed, the tumor returned in 2005, and caused Heyward's death in May 2006.
Pro skateboarder Ray Underhill, a member of the Powell-Peralta Bones Brigade, battled chordoma for two years before succumbing to his disease in August 2008.
Cary Tennis, the popular advice columnist for Salon, announced in his column of November 19, 2009, that he had been diagnosed with a chordoma.
Former Spanish footballer José Enrique was diagnosed with chordoma in May 2018 and underwent surgery to remove the tumour in June of that year. He announced in April 2019 that he had been given the all clear.
References
- National Cancer Institute (February 27, 2019). "Chordoma".
- "Primary Malignant Bone Tumors: Tumors of Bones and Joints: Merck Manual Professional". Retrieved 2009-01-04.
- Senne J, Nguyen V, Staner D, Stensby JD, Bhat AP (January 2021). "Demystifying Sacral Masses: A Pictorial Review". The Indian Journal of Radiology & Imaging. 31 (1): 185–192. doi:10.1055/s-0041-1729766. PMC 8299490. PMID 34316126.
- Walcott BP, Nahed BV, Mohyeldin A, Coumans JV, Kahle KT, Ferreira MJ (2012). "Chordoma: current concepts, management, and future directions". Lancet Oncol. 13 (2): e69–76. doi:10.1016/S1470-2045(11)70337-0. PMID 22300861.
- Lee-Jones L, Aligianis I, Davies PA, et al. (September 2004). "Sacrococcygeal chordomas in patients with tuberous sclerosis complex show somatic loss of TSC1 or TSC2" (PDF). Genes Chromosomes Cancer. 41 (1): 80–5. doi:10.1002/gcc.20052. PMID 15236319. S2CID 13136963.
- Han S, Polizzano C, Nielsen GP, Hornicek FJ, Rosenberg AE, Ramesh V (March 2009). "Aberrant Hyperactivation of Akt and Mammalian Target of Rapamycin Complex 1 Signaling in Sporadic Chordomas". Clinical Cancer Research. 15 (6): 1940–6. doi:10.1158/1078-0432.CCR-08-2364. PMC 2701205. PMID 19276265.
- Presneau N, Shalaby A, Idowu B, Gikas P, Cannon SR, Gout I, Diss T, Tirabosco R, Flanagan AM (May 2009). "Potential therapeutic targets for chordoma: PI3K/AKT/TSC1/TSC2/mTOR pathway". British Journal of Cancer. 100 (9): 1406–14. doi:10.1038/sj.bjc.6605019. PMC 2694420. PMID 19401700.
- Fasig JH, Dupont WD, LaFleur BJ, Olson SJ, Cates JM (February 2008). "Immunohistochemical analysis of receptor tyrosine kinase signal transduction activity in chordoma". Neuropathology and Applied Neurobiology. 34 (1): 95–104. doi:10.1111/j.1365-2990.2007.00873.x. PMID 17973908. S2CID 22858447.
- Hallor KH, Staaf J, Jönsson G, Heidenblad M, Vult von Steyern F, Bauer HC, Ijszenga M, Hogendoorn PC, Mandahl N, Szuhai K, Mertens F (January 2008). "Frequent deletion of the CDKN2A locus in chordoma: analysis of chromosomal imbalances using array comparative genomic hybridisation". British Journal of Cancer. 98 (2): 434–42. doi:10.1038/sj.bjc.6604130. PMC 2361468. PMID 18071362.
- Schwab JH, Boland PJ, Agaram NP, Socci ND, Guo T, O'Toole GC, Wang X, Ostroumov E, Hunter CJ, Block JA, Doty S, Ferrone S, Healey JH, Antonescu CR (March 2009). "Chordoma and chondrosarcoma gene profile: implications for immunotherapy". Cancer Immunology, Immunotherapy. 58 (3): 339–49. doi:10.1007/s00262-008-0557-7. PMC 3426285. PMID 18641983.
- "Gene Duplication Identified in an Uncommon Form of Bone Cancer". 2009. Archived from the original on 2009-10-09. Retrieved 2009-10-09.
- Wang, Ke; Zhen, Wu; Tian, Kaibing; Hao, Shuyu; Zhang, Liwei; Zhang, Junting (November 2015). "Familial Chordoma: a case report and review of the literature". Oncology Letters. Oncology Letters 10(5). 10 (5): 2937–2940. doi:10.3892/ol.2015.3687. PMC 4665336. PMID 26722267.
- "First clinical guidelines for chordoma treatment published in The Lancet Oncology". 2015-02-19.
- Chugh R, Tawbi H, Lucas DR, Biermann JS, Schuetze SM, Baker LH (November 2007). "Chordoma: the nonsarcoma primary bone tumor". The Oncologist. 12 (11): 1344–50. doi:10.1634/theoncologist.12-11-1344. hdl:2027.42/139965. PMID 18055855. S2CID 34916915.
- Park L, Delaney TF, Liebsch NJ, Hornicek FJ, Goldberg S, Mankin H, Rosenberg AE, Rosenthal DI, Suit HD (2006). "Sacral chordomas: Impact of high-dose proton/photon-beam radiation therapy combined with or without surgery for primary versus recurrent tumor". Int J Radiat Oncol Biol Phys. 65 (5): 1514–21. doi:10.1016/j.ijrobp.2006.02.059. PMID 16757128.
- Delaney TF, Liebsch NJ, Pedlow FX, Adams J, Dean S, Yeap BY, McManus P, Rosenberg AE, Nielsen GP, Harmon DC, Spiro IJ, Raskin KA, Suit HD, Yoon SS, Hornicek FJ (2009). "Sacral chordomas: Phase II Study of High-Dose Photon/Proton Radiotherapy in the Management of Spine Sarcomas". Int J Radiat Oncol Biol Phys. 74 (3): 732–9. doi:10.1016/j.ijrobp.2008.08.058. PMC 2734911. PMID 19095372.
- Casali PG, Messina A, Stacchiotti S, et al. (2004). "Imatinib mesylate in chordoma" (PDF). Cancer. 101 (9): 2086–97. doi:10.1002/cncr.20618. hdl:2434/642716. PMID 15372471.
- Verma, S., Vadlamani, S.P., Shamim, S.A. Rastogi S et al. Partial response to erlotinib in a patient with imatinib-refractory sacral chordoma. Clin Sarcoma Res 10, 28 (2020). https://doi.org/10.1186/s13569-020-00149-1
- Gröschel S, Hübschmann D, Raimondi F, Horak P, Warsow G, Fröhlich M, Klink B, Gieldon L, Hutter B, Kleinheinz K, Bonekamp D, Marschal O, Chudasama P10,11, Mika J, Groth M, Uhrig S, Krämer S, Heining C, Heilig CE, Richter D, Reisinger E, Pfütze K, Eils R, Wolf S, von Kalle C, Brandts C, Scholl C, Weichert W, Richter S, Bauer S, Penzel R, Schröck E, Stenzinger A, Schlenk RF, Brors B, Russell RB, Glimm H, Schlesner M, Fröhling S (2019) Defective homologous recombination DNA repair as therapeutic target in advanced chordoma. Nat Commun 10(1):1635
- Fuchs B, Dickey ID, Yaszemski MJ, Inwards CY, Sim FH (2005). "Operative management of sacral chordoma". The Journal of Bone and Joint Surgery. American Volume. 87 (10): 2211–6. doi:10.2106/JBJS.D.02693. PMID 16203885.
- "College student fights his own cancer - Yahoo! News". Archived from the original on 2008-02-26. Retrieved 2008-02-20.
- Kelley MJ, Shi J, Ballew B, Hyland PL, Li WQ, Rotunno M, Alcorta DA, Liebsch NJ, Mitchell J, Bass S, Roberson D, Boland J, Cullen M, He J, Burdette L, Yeager M, Chanock SJ, Parry DM, Goldstein AM, Yang XR (2014). "Familial Chordoma Study of the T Gene". Hum Genet. 133 (10): 1289–97. doi:10.1007/s00439-014-1463-z. PMC 6938388. PMID 24990759.
- "Familial Chordoma Study". Archived from the original on 2009-02-14. Retrieved 2009-02-03.
- Expert Recommendations for the Diagnosis and Treatment of Chordoma
External links
- Images of Chordoma - mostly radiological (CT and MRI scans), one autopsy image
- Research information on chordoma (WikiGenes)