Collapsin response mediator protein family
Collapsin response mediator protein family or CRMP family consists of five intracellular phosphoproteins (CRMP-1, CRMP-2, CRMP-3, CRMP-4, CRMP-5) of similar molecular size (60–66 kDa) and high (50–70%) amino acid sequence identity. CRMPs are predominantly expressed in the nervous system during development and play important roles in axon formation from neurites and in growth cone guidance and collapse through their interactions with microtubules.[1][2] Cleaved forms of CRMPs have also been linked to neuron degeneration after trauma induced injury.[3]
The modulation of CRMP-2 expression through various pharmaceuticals is a new and expanding area of research. By discovering chemicals that can either increase or decrease CRMP-2 expression, scientists can potentially reduce the effects of neurological diseases such as Alzheimer's disease and Parkinson's disease.[4][5]
History
Members of the CRMP family were discovered independently in different species by several groups working in parallel.[4][6] Among the five members of the family, CRMP-2 was first identified in 1995. Group of researchers led by Goshima found out that CRMP-2 played a role in the transduction of the extracellular Semaphorin 3A (Sema3A), an inhibitory protein for axonal guidance in chick dorsal root ganglion (DRG).[6] The protein was first named as CRMP-62 having a relative molecular mass of 62 kDa and later referred as CRMP-2. Concurrently, a 64 kDa protein named as TOAD-64 for Turned On After Division, was shown to increase significantly during the development of the cortex of the brain. The cDNA sequence of TOAD-64 corresponded to that of rat CRMP-2. In 1996, mouse CRMP-4, often referred to as Ulip for Unc-33 like phosphoprotein, was discovered by Byk and colleagues, using a rabbit polyclonal antiserum which recognized a 64 kDa mouse brain specific phosphoprotein.[6] In the same year, several other studies cloned CRMPs-1-4 in rat and dihydropyrimidinase (DHPase) homologous sequence of CRMPs-1, -2, and -4 in human fetal brain.[6] Finally, in 2000, CRMP-5 was discovered using two-hybrid screenings of brain libraries or purification from a proteic complex.[6] In following researches, CRMPs were studied as target antigens for autoantibodies in various autoimmune neurodegenerative disorders.[6]
Structure
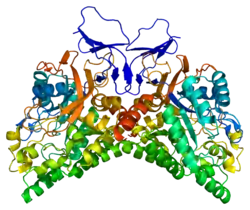
CRMP1-5 are between 564 and 572 amino acids and these proteins are found to be approximately 95% conserved between mouse and human.[6] The protein sequence of CRMP1-4 is approximately 75% homologous with each other, while CRMP5 is only 50-51% homologous with each of the other CRMPs.[4] Additionally, CRMPs are homologs of Unc-33 whose mutation causes impaired ability to form neural circuits and uncoordinated mobility in Caenorhabditis elegans.[7] CRMP1-4 genes are roughly 60% homologous with the tetramer liver dihydropyrimidinase (DHPase), and also possess a similar structure to members of the metal-dependent amidohydrolases. However, the fact that CRMPs are not enzymatic reveals that they might lack the critical His residues that are present in amidohydrolase enzymes to allow them to bind metal atoms to their active site.[4]
Additionally, CRMPs can exist as homotetramers or as heterotetramers. The tetramers are positioned so that the active residues on the N-terminal are located on the outside of the complex. This allows CRMP to regulate various factors in the cytoplasm. Gel filtration analysis has shown that CRMP-5 and CRMP-1 form weaker homo-tetramers compared with CRMP-2, and that divalent cations, Ca2+ and Mg2+, destabilize oligomers of CRMP-5 and CRMP-1, but promote CRMP-2 oligomerization.[8] The C-terminus consists of 80 amino acids and is the site of phosphorylation for various kinases.[4]
Expression
The expression of CRMPs is regulated throughout development of the nervous system. In general, CRMPs are highly expressed in post-mitotic nerve cells since early embryonic life. In the developing nervous system, each CRMP displays a distinct expression pattern both in time and space. For example, in the external granular layer (EGL), where mitosis of cerebellar granular neuron occurs, CRMP-2 is highly expressed while CRMP-5 is never expressed. However, CRMP-2 and CRMP-5 are found to be co-expressed in post-mitotic granular neurons.[6] CRMP expression is highest when neurons and synaptic connections mature actively during the first postnatal week, suggesting CRMPs’ role in neuronal migration, differentiation and axonal growth.[4][6] Indeed, CRMP-2 expression is induced by neuronal differentiation promoting factors such as noggin, chordin, GDNF, and FGF.[4]
In the adult nervous system, CRMP expression is significantly downregulated and limited in areas associated with brain plasticity, neurogenesis, or regeneration. CRMP1 mRNA is mainly expressed in Purkinje cells of the cerebellum. Among the five members of the CRMP family, CRMP-2 is the most highly expressed in the adult brain, especially in post-mitotic neurons of the olfactory system, cerebellum, and hippocampus. CRMP-3 mRNA is only expressed in the granular layer of the cerebellum, inferior olive, and dentate gyrus of the hippocampus. CRMP-4 is the least expressed protein of CRMP family and its expression is restricted to the olfactory bulb, hippocampus, and the internal granule layer (IGL) of the cerebellum. Lastly, CRMP-5 is expressed not only in post-mitotic neurons of the olfactory bulb, olfactory epithelium, and dentate gyrus of the hippocampus, but also in peripheral nerve axons and sensory neurons. Other families of CRMP also appear in peripheral tissues. Expression of CRMPs-1, -4, and -5 in the adult testis is detected only in the cell spermatid stage and CRMP-2 mRNA is found in lung tissue of the fetal mouse and adult human.[6]
The expression of CRMPs also can be found in the death or survival signaling of postmitotic neurons. Although CRMP is a cytosolic protein, significant amount of CRMP expression is detected as membrane associated at the leading edge of the growth cone lamellipodium and filopodia. Also, injury-induced CRMPs expression is found in sprouting fibers in both the central and peripheral nervous system.[4] CRMP-4 expression is promoted upon ischemic injury and is associated with neurons having intact morphology, suggesting that CRMP-4 provides a survival signal and may be involved in regeneration of neurons. Similarly, CRMP-2 has been suggested to participate in the survival and maintenance in postmitotic neurons as its over-expression accelerates nerve regeneration. However, CRMP-2 may also be involved in neuronal death as its expression is upregulated during the early stages of dopamine-induced neuronal apoptosis in cerebellar granule neurons.[7]
Mechanism, Function and Regulation
Axonal formation in developing neuron
CRMP-2 plays a role in neuronal polarity. Extensions of early neurons called lamellipodia form the early neurites. The neurites are indistinguishable between dendrites and the axon during this stage. One of these neurites eventually becomes the axon and grows longer than the dendritic neurites. CRMP-2 helps facilitate the rate of this axonal growth through its interactions with microtubules.[1] CRMP-2 binds to and copolymerizes with tubulin heterodimers but does not bind as well to polymerized tubulin. This binding specificity promotes tubulin polymerization in vitro. CRMP-2/tubulin complexes are found in the distal part of the axon and modulate microtubule dynamics by controlling the rate of microtubule assembly. CRMP-2 also contributes to the establishment of neuronal polarity by regulating polarized Numb-mediated endocytosis at the axonal growth cones.[1] In both cases, phosphorylation of CRMP-2 at Thr-555 by Rho kinase or at Thr-509, Thr-514 or Ser-518 by GSK-3β inactivates the protein by lowering binding affinity to tubulin and Numb.[1]
Axonal growth cone guidance
In the developing nervous system, CRMPs’ involvement in axonal guidance has been proposed by localization of CRMPs in neurites and axonal growth cones. CRMPs participate in two distinct transduction pathways inducing axonal growth cone collapse. Both pathways involve Rho family GTPases, RhoA and Rac1, in their signaling cascade. Rho family GTPases regulate the cytoskeletal reorganization of the growth cone and affect the growth cone motility.[2]
In Sema3A signaling cascade, CRMP plays a role as intracellular messenger mediating repulsive signal. Sema3A initiates clustering of the receptor neuropilin 1 and plexin A1.[6] While some of the other class of semaphorins directly bind to plexin receptors, Sema3A does not bind to plexin directly. Instead, it interacts with neuropilins as ligand-binding co-receptor for plexin and releases plexin-based signaling. The signal transduction pathway downstream of activated plexin receptor is mediated by CRMPs.[2] In response to Sema3A signaling cascade, CRMPs which exist as a heterotetramer in the cytosol bind to the cytosolic domain of PlexA and its conformation changes. Further, CRMPs are phosphorylated by Cdk5, GSK3B, and Fes, a tyrosine protein kinase.[4] Especially, phosphorylation of CRMP-1 and CRMP-2 are essential for Sema3A-regulated axonal guidance.[7] In the presence of CRMP-2, the signal can induce alterations of Rac-dependent pathway, which modulates the actin filament assembly in the growth cone. In the absence of Sema3A, the interaction between CRMP tetramer and PlexA is blocked.[4] Phospholipase D2 (PLD-2) which is localized in the growth cone and is involved in actin cytoskeleton rearrangement, can be inhibited by CRMP-2 and its inhibition results in actin depolymerization and possibly affects axonal growth cone collapse. In the presence of CRMP-2, the signal can induce alterations of Rac-dependent pathway, which modulates the actin filament assembly in the growth cone.[6]
CRMP-2 is also involved in another growth cone collapse signal induced by extracellular lysophosphatidic acid (LPA). A signal through seven-transmembrane receptor activates an intracellular pathway, RhoA and the downstream of RhoA, Rho-kinase subsequently phosphorylates CRMP-2 on Threonine-555 (Thr555). In DRG neurons, CRMP-2 is phosphorylated by Rho kinase in LPA signaling but not in Sema3A signaling, revealing the presence of both Rho kinase-dependent and Rho kinase-independent pathways for the growth cone collapse.[2] In RhoA pathway, CRMP-1 interacts with Rho-kinase and modulates RhoA signaling. CRMP-2 can be regulated post-translationally by O-GluNAc (β-N-acetylglucosamine linked to hydroxyls of serine or threonine) as the modification blocks CRMP-2 from being phosphorylated.[6]
Trauma induced degeneration
Cleaved CRMP products play a considerable role in the degeneration of axons as a result of trauma inflicted on the central nervous system (CNS). As a result of trauma induced on the CNS, glutamate activates NMDA receptors leading to an influx of calcium that activates the calcium-dependent protease calpain. It has been shown that activated calpain proteolytically cleaves CRMP-3, creating a cleavage product of CRMP that interacts with vital cytosolic and nuclear molecules to bring about neurodegeneration.[7] The structure of this cleaved form of CRMP has not been determined yet, making it difficult to understand the protein-protein interactions that occur and why these forms are able to initiate neurodegeneration after CNS injury. Additionally, calpain inhibitors (ALLN) are shown to have prevented the CRMP‐3 cleavage and therefore no axonal degeneration or neuronal death, further suggesting that calpain targets CRMP-3 for cleavage during glutamate-induced neuronal death. Ca2+/calmodulin-dependent protein kinase II (CaMK II) is also activated by calcium influx through NMDA receptors, and is another possible activator of CRMP-3.[7] CRMP-3 is not the only CRMP involved in neuronal degeneration brought upon by trauma and cerebral ischemia, as all CRMPs are in fact targeted for cleavage to help promote degeneration.[7]
List of CRMPs (and associated knockout phenotypes and derived functions)
| CRMP | Phenotype in knockout mice | Derived Function in cultured neurons |
|---|---|---|
| CRMP-1 | Decrease in granule cell proliferation and apoptosis | Sem3A-induced axonal guidance effect |
| Retarded neuronal migration | Axon formation/extension induced by NT3 | |
| Disorientation of apical dendrites | Death of spinal cord neurons | |
| Impaired dendritic spine density | ||
| Impaired LTP and spatial memory | ||
| CRMP-2 | Severely abnormal dendritic patterning | Axonal guidance by chemorepellent |
| Axon specification, elongation and branching | ||
| NT3-induced axon outgrowth | ||
| Negative effect on axon extension induced by NGF | ||
| Accelerates axon regeneration of nerve-injured motor neurons | ||
| Neurotransmitter release | ||
| Resistance to glutamate toxicity through NR2B trafficking | ||
| Neuronal cell death | ||
| CRMP-3 | Impaired dendritic spine maturation | Neuronal cell death |
| Impaired LTP | ||
| Decrease of prepulse inhibition | ||
| CRMP-4 | Increased proximal bifurcation phenotype in the CA1 hippocampus | Axon elongation and branching |
| Inhibition of axon regeneration by myelin-derived inhibitors | ||
| Axonal degeneration and cell death | ||
| Sema3A-induced extension and branching of dendrites | ||
| CRMP-5 | Atrophy of Purkinje cells | Filopodia and growth cone development |
| Impaired LTD | Abrogation of the neurite outgrowth promotional activity of CRMP-2 |
Clinical significance
The expression of CRMPs is altered in neurodegenerative diseases and these proteins likely play an essential role in the pathogenesis of disorders in the nervous system, including Alzheimer's disease, Parkinson's disease, schizophrenia, and many others. One pharmaceutical that is relatively effective in targeting CRMP-2 to reduce the results of a neurodegenerative disease is lacosamide. Lacosamide is used in combination with other types of medications to control various types of seizures, especially epilepsy. One of the ways lacosamide does this is by modulating CRMP-2, thus inducing neuroprotective effects and decreasing the epileptic effects in people with epilepsy.[10]
CRMP-2 phosphorylated at Thr-509, Ser-518, and Ser-522 has been connected to the degenerating neuritis in Alzheimer's disease. Studies suggest that glycogen synthase kinase-3β (GSK-3β) and cyclin-dependent protein kinase 5 (Cdk5) are highly expressed in Alzheimer's disease and are some of the protein kinases responsible for inactivating CRMP-2 in Alzheimer's disease. This inactivation of CRMP-2 in people with Alzheimer's disease promotes the expression of neurofibrillary tangles and plaque neurites which are consistent with people with this disease.[11][12] CRMP-2 is also related to bipolar disorder and schizophrenia, likely as a result of the phosphorylation of CRMP-2 by GSK-3β.[12]
References
- Arimura N, Menager C, Fukata Y, Kaibuchi K (January 2004). "Role of CRMP-2 in neuronal polarity". Journal of Neurobiology. 58 (1): 34–47. doi:10.1002/neu.10269. PMID 14598368.
- Liu BP, Strittmatter SM (October 2001). "Semaphorin-mediated axonal guidance via Rho-related G proteins". Current Opinion in Cell Biology. 13 (5): 619–626. doi:10.1016/s0955-0674(00)00260-x. PMID 11544032.
- Taghian K, Lee JY, Petratos S (August 2012). "Phosphorylation and Cleavage of the Family of Collapsin Response Mediator Proteins May Play a Central Role in Neurodegeneration after CNS Trauma". Journal of Neurotrauma. 29 (9): 1728–1735. doi:10.1089/neu.2011.2063. PMID 22181040.
- Schmidt EF, Strittmatter SM (2007). "The CRMP Family of Proteins and Their Role in Sema3A Signaling". Semaphorins: Receptor and Intracellular Signaling Mechanisms. Advances in Experimental Medicine and Biology. Vol. 600. pp. 1–11. doi:10.1007/978-0-387-70956-7_1. ISBN 978-0-387-70955-0. PMC 2853248. PMID 17607942.
- Quach TT, Moutal A, Khanna R, Deems NP, Duchemin AM, Barrientos RM (2020). "Collapsin Response Mediator Proteins: Novel Targets for Alzheimer's Disease". J Alzheimers Dis. 77 (3): 949–960. doi:10.3233/JAD-200721. PMC 7579750. PMID 32804096.
- Charrier E.; Reibel S.; Rogemond V.; Aguera M.; Thomasset N.; Honnorat J. (August 2003). "Collapsin response mediator proteins (CRMPs) - Involvement in nervous system development and adult neurodegenerative disorders". Molecular Neurobiology. 28 (1): 51–63. doi:10.1385/MN:28:1:51. PMID 14514985. S2CID 23916946.
- Hou ST, Jiang SX, Smith RA (2008). Permissive and repulsive cues and signaling pathways of axonal outgrowth and regeneration. pp. 125–181. doi:10.1016/S1937-6448(08)00603-5. ISBN 9780123743749. PMID 18544498.
{{cite book}}:|journal=ignored (help) - Ponnusamy, Rajesh; Bernhard Lohkamp (5 March 2013). "Insights into the oligomerization of CRMPs: crystal structure of human collapsin response mediator protein 5". Journal of Neurochemistry. DOI: 10.1111/jnc.12188. 125 (6): 855–868. doi:10.1111/jnc.12188. PMID 23373749. S2CID 22590453.
- Yamahista N, Goshima Y (February 2012). "Collapsin Response Mediator Proteins Regulate Neuronal Development and Plasticity by Switching Their Phosphorylation Status". Molecular Neurobiology. 45 (1): 234–246. doi:10.1007/s12035-012-8242-4. PMID 22351471. S2CID 17567061.
- Doty P, Rudd GD, Stoehr T, Thomas D (January 2007). "Lacosamide". Neurotherapeutics. 4 (1): 145–8. doi:10.1016/j.nurt.2006.10.002. PMC 7479700. PMID 17199030.
- Soutar MP, Thornhill P, Cole AR, Sutherland C (June 2009). "Increased CRMP2 phosphorylation is observed in Alzheimer's disease; does this tell us anything about disease development?". Current Alzheimer Research. 6 (3): 269–78. doi:10.2174/156720509788486572. PMID 19519308.
- Yoshimura T, Arimura N, Kaibuchi K (November 2006). "Molecular mechanisms of axon specification and neuronal disorders". Annals of the New York Academy of Sciences. 1086 (1): 116–125. Bibcode:2006NYASA1086..116Y. doi:10.1196/annals.1377.013. PMID 17185510. S2CID 37627165.