Double-strand break repair model
A double-strand break repair model refers to the various models of pathways that cells undertake to repair double strand-breaks (DSB). DSB repair is an important cellular process, as the accumulation of unrepaired DSB could lead to chromosomal rearrangements, tumorigenesis or even cell death.[1] In human cells, there are two main DSB repair mechanisms: Homologous recombination (HR) and non-homologous end joining (NHEJ). HR relies on undamaged template DNA as reference to repair the DSB, resulting in the restoration of the original sequence.[2] NHEJ modifies and ligates the damaged ends regardless of homology.[2] In terms of DSB repair pathway choice, most mammalian cells appear to favor NHEJ rather than HR. This is because the employment of HR may lead to gene deletion or amplification in cells which contains repetitive sequences.[1] In terms of repair models in the cell cycle, HR is only possible during the S and G2 phases, while NHEJ can occur throughout whole process.[3] These repair pathways are all regulated by the overarching DNA damage response mechanism.[4] Besides HR and NHEJ, there are also other repair models which exists in cells. Some are categorized under HR, such as synthesis-dependent strain annealing, break-induced replication, and single-strand annealing; while others are an entirely alternate repair model, namely, the pathway microhomology-mediated end joining (MMEJ).[5]
Causes
DSB can occur naturally due to the presence of reactive species generated by metabolism, and various external factors (e.g. ionizing radiation or chemotherapeutic drugs).[1]
In mammalian cells, there are numerous cellular processes that induce DSB. Firstly, DNA topological strain from topoisomerase during normal cell growth can cause the majority a cell’s DSB.[6] Secondly, cellular processes such as meiosis and the maturation of antibodies can cause nuclease-induced DSB.[7] Thirdly, the cleavage of different DNA structures such as reversed or blocked DNA replication forks, R-loops and DNA interstrand crosslinks can also cause DSB.[7]
Different models
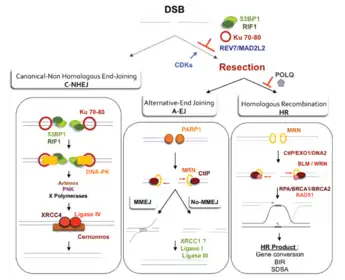
Homologous recombination
Homologous recombination involves the exchange of DNA materials between homologous chromosomes. There are multiple pathways of HR to repair DSBs, which includes double-strand break repair (DSBR), synthesis-dependent strand annealing (SDSA), break-induced replication (BIR), and single-strand annealing (SSA).[8]
The regulation of HR in mammalian cells involves key HR proteins such as BRCA1 and BRCA2.[9] And as mentioned, since HR can lead to aggressive chromosomal rearrangement, loss of genetic information that could contribute to cell death, it explains why HR is strictly regulated.[8]
Double-strand break repair

HR repairs DSB by copying intact and homologous DNA molecules. The blunt ends of the DSB are processed into ssDNA with 3’ extensions, which allows RAD51 recombinase (eukaryotic homologue of prokaryotic RecA) to bind to it to form a nucleoprotein filament.[3][10] The function of the filament is to locate the template DNA and form a joint heteroduplex molecule. Other proteins such as RP-A protein and RAD52 also coordinate in the heteroduplex formation,[10] the RP-A protein has to be removed for the RAD51 to form the filament,[11] whereas the RAD52 is a key HR mediator.[3] Afterwards, the 3’ ssDNA invades the template DNA, and displaces a DNA strand to form a D-loop. DNA polymerase and other accessory factors follows by replacing the missing DNA via DNA synthesis. Ligase then attaches the DNA strand break,[10] resulting in the formation of 2 Holliday junctions. The recombined DNA strands then undergoes resolution by cleavage. The orientation of the cleavage determines whether the resolution results in either cross-over or noncross-over products.[12] Lastly, the strands finally separate and revert to its original form.
, the main pathway for resolution relies on the BTR (BLM helicase-TopoisomeraseIIIα-RMI1-RM2) complex, where it induces the resolution of the 2 Holliday junctions, but this pathway favors the noncross-over cleavage.[12]
Synthesis-dependent strain annealing
Synthesis-dependent strain annealing is the most preferred repair mechanism in somatic cells.[3] The pathway of SDSA is similar to DSBR until just after the D-loop formation. Instead of forming Holliday junctions after DNA synthesis, the nascent strand dissociates via RETL1 helicase and anneals back to the other end of the resected strand.[3][9][13] This explains why SDSA results in a non-crossover pathway.[3] The remaining gap is filled in and the nick is attached by the ligase.[9]
Break-induced replication
Although there is little research in regards of break-induced replication, it is known that it is a one-ended recombination mechanism, where only of the one ends of a DSB will be involved in strand invasion.[14] This means that unlike DSBR, BIR does not link back to the second DSB end after the strand invasion and replication.[14]
Single-strand annealing
Single-strand annealing involves homologous/repeated sequences flanking a DSB.[7] The process starts with the key end resection factor CtlP, which mediates the end resection of DSBs, resulting in the formation of a 3' ssDNA extension. Meditated by RAD52, the flanking homologous sequences are annealed, and forms a synapse intermediate.[7] Then, the nonhomologous 3’ extension is removed by the ERCC1-XPF complex through endonucleolytic cleavage, with RAD52 increasing the efficiency of the ERCC1-XPF complex activity.[7] It is only after the removal of 3’ ssDNA, where the polymerase will fill the missing gaps and the ligase to ligate the strands.[7] Since SSA results in the deletion of repetitive sequences, this could potentially lead to error-prone repair.[3]
Single-strand annealing differs from SDSA and DSBR in numerous ways. For instance, the 3’ extension after the end resection in SSA anneals to the repeated/homologous sequences of the other end, whereas in other pathways the strand invasion to another homologous DNA template.[15] Moreover, SSA does not require RAD51, because it does not involve strand invasion, but rather the annealing of homologous sequences.[3]
Non-homologous end joining
Non-homologous end joining (NHEJ) is one of the major pathways in DSB repair besides HR.[16] The basic concept of NHEJ involves three steps. First, the ends of a DSB is captured by a group of enzymes. The enzymes then form a bridge which connects the DSB ends together, and is lastly followed by religation of the DNA strands.[17] To initiate whole process, the Ku70/80 protein complex binds to the damaged ends of the DSB strands. This forms a preliminary scaffold which allows the recruitment of various NHEJ factors, such as the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), DNA Ligase IV and X-ray cross complementing protein 4 (XRCC4) to form a bridge and bring both ends of the damaged DNA strands together.[18][19][20][21] This is then followed by the processing of any non-ligatable DNA termini by a group of proteins including Artemis, PNKP, APLF and Ku, before the XRCC4 and DNA Ligase IV ligate the bridged DNA.[17][22]
Microhomology-mediated end joining
Microhomology-mediated end joining (MMEJ), also known as alt-non-homologous end joining, is another pathway to repair DSBs. The process of MMEJ can be summarized in five steps: the 5' to 3' cutting of DNA ends, annealing of microhomology, removing heterologous flaps, and ligation and synthesis of gap filling DNA.[5] It was found that the selection between MMEJ and NHEJ is mainly dependent on Ku levels and the concurrent cell cycle.[23]
The regulation of double-strand break repair pathways
DNA damage response
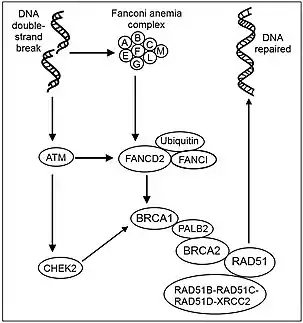
DNA damage response (DDR) is the overarching mechanism which mediates the cell's detection and response to DNA damage. This includes the process of detecting DSB within the cell, and the subsequent triggering and regulation of DSB repair pathways. Upstream detections of DNA damage via DDR will lead to the activation of downstream responses such as senescence, cell apoptosis, halting transcription and activating DNA repair mechanisms.[4] Proteins such as the proteins ATM, ATR and DNA-dependent protein kinase (DNA-PK) are vital for the process of detection of DSB in DDR, and these proteins are recruited to the DSB site in the DNA.[24] In particular, ATM has been identified as the protein kinase in charge of the global meditation of cellular responses to DSB, which includes various DSB repair pathways.[24] Following the recruitment of the aforementioned proteins to DNA damage sites, they will in turn trigger cellular responses and repair pathways to mitigate and repair the damage caused.[4] In short, these vital upstream proteins and downstream repair pathways altogether forms the DDR, which plays a vital role in DSB repair pathways regulation.
Double-strand break repair pathway choice
As cells have developed various DSB repair models, it is said that specific pathways are favoured for their ability to repair DSB depending on the cellular context.[25] These conditions include the type of DSB involved, the species of cells involved, and the stage of the cell cycle.[26]
In various types of DSB
Cells have evolved a multitude of DSB repair pathways in response to the various types of DSB.[26] Hence, various pathways are favoured in different situations. For instance, frank DSB, which are DSB induced by substances like as ionizing radiation, and nucleases, can be repaired by both HR and NHEJ. On the other hand, DSB due to replication fork collapse mainly favours HR.[26][27]
In higher eukaryotes and yeast cells
It is said that the favoured pathway in a particular situations is also largely dependent on the species of the cell, the cell type, and cell cycle phases; and are all modulated and triggered by different upstream regulatory proteins.[26] As compared to higher eukaryotes, yeast cells have adopted HR as the main repair pathway for DSB.[28] Imprecise NHEJ, the primary pathway for NHEJ to repair "dirty" ends due to IR, was found to be inefficent at repairing DSB in yeast cells. It was hypothesized that this inefficiency as compared to mammalian cells is due to the lack of three vital NHEJ proteins, including DNA-PKcs, BRCA1, and Artemis.[26] Contrary to yests, higher eukaryotes has a much higher frequency and efficiency at adopting NHEJ pathways.[29] Research hypothesize that this is due to the higher eukaryote's larger genome size, as it means that more NHEJ related proteins are encoded for NHEJ repair pathways; and a larger genome implies a challenging obstacle to find a homologous template for HR.[26]
In cell cycle
HR and NHEJ pathways are favoured in various phases of cell cycles for a multitude of factors. As S and G2 phases of the cell cycle generate more chromatids, the increased availability of template access for HR results in the up-regulation of the pathway.[30] This rise is further increased due to the activation of CDK1 and the increase of RAD51 and RAD52 levels during G1 phase.[26][31] Despite this, NHEJ not is inactive during the HR up-regulation. In fact, NHEJ was shown to be active throughout all stages of the cell cycle, and is favoured in G1 phase during low resection action intervals.[32][33] This suggests the competition between HR and NHEJ for DSB repair in cells.[31] It should be noted, however, that there is a shift of favour from NHEJ to HR when the cell cycle is progressing from G1 to S/G2 phases in eukaryotic cells.[31]
Defective DSB repair
Although there is no universal model to explain disease etiology caused by DNA repair deficiency, it is said that the accumulation of unrepaired DNA damage may lead to various diseases, including various metabolic syndromes and types of cancers.[34][35] Some examples of diseases caused by defects of DSB repair mechanisms are listed below:
- Fanconi Anemia (FA) and Hereditary breast and ovarian cancer (HBOC) syndrome are caused by defects in homologous recombination.[36] Biallelic mutation of either BRCA1/2 gene results in the loss of homologous recombination activity.[36]
- Chordomas, a rare bone tumour, might suggest defects in homologous recombination and mutations affecting HR-related genes.[37]
- Defects in the NHEJ mechanism are related to the mutations in hRAD50 and/or hMRE11 genes in mismatch repair deficient tumors.[38]
See also
References
- Featherstone C, Jackson SP (October 1999). "DNA double-strand break repair". Current Biology. 9 (20): R759–R761. doi:10.1016/S0960-9822(00)80005-6. PMID 10531043. S2CID 1941783.
- Mao Z, Bozzella M, Seluanov A, Gorbunova V (October 2008). "Comparison of nonhomologous end joining and homologous recombination in human cells". DNA Repair. 7 (10): 1765–1771. doi:10.1016/j.dnarep.2008.06.018. PMC 2695993. PMID 18675941.
- Scully R, Panday A, Elango R, Willis NA (November 2019). "DNA double-strand break repair-pathway choice in somatic mammalian cells". Nature Reviews. Molecular Cell Biology. 20 (11): 698–714. doi:10.1038/s41580-019-0152-0. PMC 7315405. PMID 31263220.
- Zhou BB, Elledge SJ (November 2000). "The DNA damage response: putting checkpoints in perspective". Nature. 408 (6811): 433–439. Bibcode:2000Natur.408..433Z. doi:10.1038/35044005. PMID 11100718. S2CID 4419141.
- Sfeir A, Symington LS (November 2015). "Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway?". Trends in Biochemical Sciences. 40 (11): 701–714. doi:10.1016/j.tibs.2015.08.006. PMC 4638128. PMID 26439531.
- Tan XY, Huen MS (October 2020). "Perfecting DNA double-strand break repair on transcribed chromatin". Essays in Biochemistry. 64 (5): 705–719. doi:10.1042/EBC20190094. PMID 32309851. S2CID 216030185.
- Bhargava R, Onyango DO, Stark JM (September 2016). "Regulation of Single-Strand Annealing and its Role in Genome Maintenance". Trends in Genetics. 32 (9): 566–575. doi:10.1016/j.tig.2016.06.007. PMC 4992407. PMID 27450436.
- Sebesta M, Krejci L (2016). "Mechanism of Homologous Recombination". In Hanaoka F, Sugasawa K (eds.). DNA Replication, Recombination, and Repair. pp. 73–109. doi:10.1007/978-4-431-55873-6_4. ISBN 978-4-431-55873-6. Retrieved 2021-03-31.
{{cite book}}:|work=ignored (help) - Do AT, Brooks JT, Le Neveu MK, LaRocque JR (March 2014). "Double-strand break repair assays determine pathway choice and structure of gene conversion events in Drosophila melanogaster". G3. 4 (3): 425–432. doi:10.1534/g3.113.010074. PMC 3962482. PMID 24368780.
- Modesti M, Kanaar R (2001-04-26). "Homologous recombination: from model organisms to human disease". Genome Biology. 2 (5): REVIEWS1014. doi:10.1186/gb-2001-2-5-reviews1014. PMC 138934. PMID 11387040.
- Wright WD, Shah SS, Heyer WD (July 2018). "Homologous recombination and the repair of DNA double-strand breaks". The Journal of Biological Chemistry. 293 (27): 10524–10535. doi:10.1074/jbc.TM118.000372. PMC 6036207. PMID 29599286.
- Shah Punatar R, Martin MJ, Wyatt HD, Chan YW, West SC (January 2017). "Resolution of single and double Holliday junction recombination intermediates by GEN1". Proceedings of the National Academy of Sciences of the United States of America. 114 (3): 443–450. Bibcode:2017PNAS..114..443S. doi:10.1073/pnas.1619790114. PMC 5255610. PMID 28049850.
- Currall BB, Chiang C, Talkowski ME, Morton CC (June 2013). "Mechanisms for Structural Variation in the Human Genome". Current Genetic Medicine Reports. 1 (2): 81–90. doi:10.1007/s40142-013-0012-8. PMC 3665418. PMID 23730541.
- Kraus E, Leung WY, Haber JE (July 2001). "Break-induced replication: a review and an example in budding yeast". Proceedings of the National Academy of Sciences of the United States of America. 98 (15): 8255–8262. Bibcode:2001PNAS...98.8255K. doi:10.1073/pnas.151008198. PMC 37429. PMID 11459961.
- Lok BH, Powell SN (December 2012). "Molecular pathways: understanding the role of Rad52 in homologous recombination for therapeutic advancement". Clinical Cancer Research. 18 (23): 6400–6406. doi:10.1158/1078-0432.CCR-11-3150. PMC 3513650. PMID 23071261.
- Radhakrishnan SK, Jette N, Lees-Miller SP (May 2014). "Non-homologous end joining: emerging themes and unanswered questions". DNA Repair. Recent Developments in Non-Homologous End Joining. 17: 2–8. doi:10.1016/j.dnarep.2014.01.009. PMC 4084493. PMID 24582502.
- Weterings E, Chen DJ (January 2008). "The endless tale of non-homologous end-joining". Cell Research. 18 (1): 114–124. doi:10.1038/cr.2008.3. PMID 18166980. S2CID 2090745.
- Uematsu N, Weterings E, Yano K, Morotomi-Yano K, Jakob B, Taucher-Scholz G, et al. (April 2007). "Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks". The Journal of Cell Biology. 177 (2): 219–229. doi:10.1083/jcb.200608077. PMC 2064131. PMID 17438073.
- Mari PO, Florea BI, Persengiev SP, Verkaik NS, Brüggenwirth HT, Modesti M, et al. (December 2006). "Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4". Proceedings of the National Academy of Sciences of the United States of America. 103 (49): 18597–18602. Bibcode:2006PNAS..10318597M. doi:10.1073/pnas.0609061103. PMC 1693708. PMID 17124166.
- Costantini S, Woodbine L, Andreoli L, Jeggo PA, Vindigni A (June 2007). "Interaction of the Ku heterodimer with the DNA ligase IV/Xrcc4 complex and its regulation by DNA-PK". DNA Repair. 6 (6): 712–722. doi:10.1016/j.dnarep.2006.12.007. PMID 17241822.
- Nick McElhinny SA, Snowden CM, McCarville J, Ramsden DA (May 2000). "Ku recruits the XRCC4-ligase IV complex to DNA ends". Molecular and Cellular Biology. 20 (9): 2996–3003. doi:10.1128/MCB.20.9.2996-3003.2000. PMC 85565. PMID 10757784.
- Davis AJ, Chen DJ (June 2013). "DNA double strand break repair via non-homologous end-joining". Translational Cancer Research. 2 (3): 130–143. doi:10.3978/j.issn.2218-676X.2013.04.02. PMC 3758668. PMID 24000320.
- Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, et al. (May 2013). "Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells". Proceedings of the National Academy of Sciences of the United States of America. 110 (19): 7720–7725. Bibcode:2013PNAS..110.7720T. doi:10.1073/pnas.1213431110. PMC 3651503. PMID 23610439.
- Blackford AN, Jackson SP (June 2017). "ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response". Molecular Cell. 66 (6): 801–817. doi:10.1016/j.molcel.2017.05.015. PMID 28622525. S2CID 206997898.
- Chapman JR, Taylor MR, Boulton SJ (August 2012). "Playing the end game: DNA double-strand break repair pathway choice". Molecular Cell. 47 (4): 497–510. doi:10.1016/j.molcel.2012.07.029. PMID 22920291.
- Shrivastav M, De Haro LP, Nickoloff JA (January 2008). "Regulation of DNA double-strand break repair pathway choice". Cell Research. 18 (1): 134–147. doi:10.1038/cr.2007.111. PMID 18157161. S2CID 20992607.
- Rothstein R, Michel B, Gangloff S (January 2000). "Replication fork pausing and recombination or "gimme a break"". Genes & Development. 14 (1): 1–10. doi:10.1101/gad.14.1.1. PMID 10640269. S2CID 40751623.
- Sugawara N, Haber JE (February 1992). "Characterization of double-strand break-induced recombination: homology requirements and single-stranded DNA formation". Molecular and Cellular Biology. 12 (2): 563–575. doi:10.1128/MCB.12.2.563. PMC 364230. PMID 1732731.
- Lin Y, Lukacsovich T, Waldman AS (December 1999). "Multiple pathways for repair of DNA double-strand breaks in mammalian chromosomes". Molecular and Cellular Biology. 19 (12): 8353–8360. doi:10.1128/MCB.19.12.8353. PMC 84924. PMID 10567560.
- Dronkert ML, Beverloo HB, Johnson RD, Hoeijmakers JH, Jasin M, Kanaar R (May 2000). "Mouse RAD54 affects DNA double-strand break repair and sister chromatid exchange". Molecular and Cellular Biology. 20 (9): 3147–3156. doi:10.1128/MCB.20.9.3147-3156.2000. PMC 85609. PMID 10757799.
- Chen F, Nastasi A, Shen Z, Brenneman M, Crissman H, Chen DJ (September 1997). "Cell cycle-dependent protein expression of mammalian homologs of yeast DNA double-strand break repair genes Rad51 and Rad52". Mutation Research. 384 (3): 205–211. doi:10.1016/S0921-8777(97)00020-7. PMID 9330616.
- Aylon Y, Liefshitz B, Kupiec M (December 2004). "The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle". The EMBO Journal. 23 (24): 4868–4875. doi:10.1038/sj.emboj.7600469. PMC 535085. PMID 15549137.
- Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, et al. (October 2004). "DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1". Nature. 431 (7011): 1011–1017. Bibcode:2004Natur.431.1011I. doi:10.1038/nature02964. PMC 4493751. PMID 15496928.
- Tiwari V, Wilson DM (August 2019). "DNA Damage and Associated DNA Repair Defects in Disease and Premature Aging". American Journal of Human Genetics. 105 (2): 237–257. doi:10.1016/j.ajhg.2019.06.005. PMC 6693886. PMID 31374202.
- Mercer JR, Cheng KK, Figg N, Gorenne I, Mahmoudi M, Griffin J, et al. (October 2010). "DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome". Circulation Research. 107 (8): 1021–1031. doi:10.1161/CIRCRESAHA.110.218966. PMC 2982998. PMID 20705925.
- Katsuki Y, Takata M (October 2016). "Defects in homologous recombination repair behind the human diseases: FA and HBOC". Endocrine-Related Cancer. 23 (10): T19–T37. doi:10.1530/ERC-16-0221. PMID 27550963.
- Gröschel S, Hübschmann D, Raimondi F, Horak P, Warsow G, Fröhlich M, et al. (April 2019). "Defective homologous recombination DNA repair as therapeutic target in advanced chordoma". Nature Communications. 10 (1): 1635. Bibcode:2019NatCo..10.1635G. doi:10.1038/s41467-019-09633-9. PMC 6456501. PMID 30967556.
- Koh KH, Kang HJ, Li LS, Kim NG, You KT, Yang E, et al. (September 2005). "Impaired nonhomologous end-joining in mismatch repair-deficient colon carcinomas". Laboratory Investigation; A Journal of Technical Methods and Pathology. 85 (9): 1130–1138. doi:10.1038/labinvest.3700315. PMID 16025146. S2CID 21197331.