ENCODE
The Encyclopedia of DNA Elements (ENCODE) is a public research project which aims "to build a comprehensive parts list of functional elements in the human genome."[2]
| Content | |
|---|---|
| Description | Whole-genome database |
| Contact | |
| Research center | Stanford University |
| Laboratory | Stanford Genome Technology Center: Cherry Lab; Formerly: University of California, Santa Cruz |
| Authors | Eurie L. Hong and 17 others[1] |
| Primary citation | PMID 26980513 |
| Release date | 2010 |
| Access | |
| Website | encodeproject |
ENCODE also supports further biomedical research by "generating community resources of genomics data, software, tools and methods for genomics data analysis, and products resulting from data analyses and interpretations."[3][4]
The current phase of ENCODE (2016-2019) is adding depth to its resources by growing the number of cell types, data types, assays and now includes support for examination of the mouse genome.[3]
History
ENCODE was launched by the US National Human Genome Research Institute (NHGRI) in September 2003.[5][6][7][8][9] Intended as a follow-up to the Human Genome Project, the ENCODE project aims to identify all functional elements in the human genome.[10]
The project involves a worldwide consortium of research groups, and data generated from this project can be accessed through public databases. The initial release of ENCODE was in 2013 and since has been changing according to the recommendations of consortium members and the wider community of scientists who use the Portal to access ENCODE data. The two-part goal for ENCODE is to serve as a publicly accessible data base for "experimental protocols, analytical procedures and the data themselves," and "the same interface should serve carefully curated metadata that record the provenance of the data and justify its interpretation in biological terms."[11] The project began its fourth phase (ENCODE 4) in February 2017.[12]
Motivation and Significance
Humans are estimated to have approximately 20,000 protein-coding genes, which account for about 1.5% of DNA in the human genome. The primary goal of the ENCODE project is to determine the role of the remaining component of the genome, much of which was traditionally regarded as "junk". The activity and expression of protein-coding genes can be modulated by the regulome - a variety of DNA elements, such as promoters, transcriptional regulatory sequences, and regions of chromatin structure and histone modification. It is thought that changes in the regulation of gene activity can disrupt protein production and cell processes and result in disease. Determining the location of these regulatory elements and how they influence gene transcription could reveal links between variations in the expression of certain genes and the development of disease.[13]
ENCODE is also intended as a comprehensive resource to allow the scientific community to better understand how the genome can affect human health, and to "stimulate the development of new therapies to prevent and treat these diseases".[6]
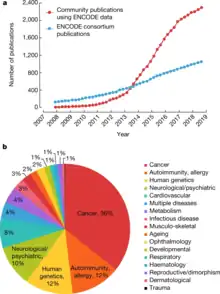
The ENCODE Consortium
The ENCODE Consortium is composed primarily of scientists who were funded by US National Human Genome Research Institute (NHGRI). Other participants contributing to the project are brought up into the Consortium or Analysis Working Group.
The pilot phase consisted of eight research groups and twelve groups participating in the ENCODE Technology Development Phase. After 2007, the number of participants expanded to 440 scientists based in 32 laboratories worldwide as the pilot phase was officially over. At the moment the consortium consists of different centers which perform different tasks.
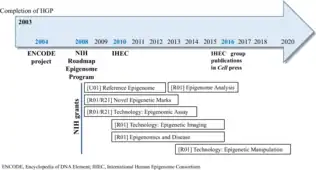
ENCODE is a member of the International Human Epigenome Consortium (IHEC).[15]
NHGRI's main requirement for the products from ENCODE-funded research is to be shared in a free, highly accessible manner to all researchers to promote genomic research. ENCODE research allows the reproducibility and thus transparency of the software, methods, data, and other tools related to the genomic analysis.[3]
The ENCODE Project
ENCODE is currently implemented in four phases: the pilot phase and the technology development phase, which were initiated simultaneously;[16] and the production phase. The fourth phase is a continuation of the third, and includes functional characterization and further integrative analysis for the encyclopedia.
The goal of the pilot phase was to identify a set of procedures that, in combination, could be applied cost-effectively and at high-throughput to accurately and comprehensively characterize large regions of the human genome. The pilot phase had to reveal gaps in the current set of tools for detecting functional sequences, and was also thought to reveal whether some methods used by that time were inefficient or unsuitable for large-scale utilization. Some of these problems had to be addressed in the ENCODE technology development phase, which aimed to devise new laboratory and computational methods that would improve our ability to identify known functional sequences or to discover new functional genomic elements. The results of the first two phases determined the best path forward for analyzing the remaining 99% of the human genome in a cost-effective and comprehensive production phase.[6]
The ENCODE Phase I Project: The Pilot Project
The pilot phase tested and compared existing methods to rigorously analyze a defined portion of the human genome sequence. It was organized as an open consortium and brought together investigators with diverse backgrounds and expertise to evaluate the relative merits of each of a diverse set of techniques, technologies and strategies. The concurrent technology development phase of the project aimed to develop new high throughput methods to identify functional elements. The goal of these efforts was to identify a suite of approaches that would allow the comprehensive identification of all the functional elements in the human genome. Through the ENCODE pilot project, National Human Genome Research Institute (NHGRI) assessed the abilities of different approaches to be scaled up for an effort to analyze the entire human genome and to find gaps in the ability to identify functional elements in genomic sequence.
The ENCODE pilot project process involved close interactions between computational and experimental scientists to evaluate a number of methods for annotating the human genome. A set of regions representing approximately 1% (30 Mb) of the human genome was selected as the target for the pilot project and was analyzed by all ENCODE pilot project investigators. All data generated by ENCODE participants on these regions was rapidly released into public databases.[8][17]
Target Selection
For use in the ENCODE pilot project, defined regions of the human genome - corresponding to 30Mb, roughly 1% of the total human genome - were selected. These regions served as the foundation on which to test and evaluate the effectiveness and efficiency of a diverse set of methods and technologies for finding various functional elements in human DNA.
Prior to embarking upon the target selection, it was decided that 50% of the 30Mb of sequence would be selected manually while the remaining sequence would be selected randomly. The two main criteria for manually selected regions were: 1) the presence of well-studied genes or other known sequence elements, and 2) the existence of a substantial amount of comparative sequence data. A total of 14.82Mb of sequence was manually selected using this approach, consisting of 14 targets that range in size from 500kb to 2Mb.
The remaining 50% of the 30Mb of sequence were composed of thirty, 500kb regions selected according to a stratified random-sampling strategy based on gene density and level of non-exonic conservation. The decision to use these particular criteria was made in order to ensure a good sampling of genomic regions varying widely in their content of genes and other functional elements. The human genome was divided into three parts - top 20%, middle 30%, and bottom 50% - along each of two axes: 1) gene density and 2) level of non-exonic conservation with respect to the orthologous mouse genomic sequence (see below), for a total of nine strata. From each stratum, three random regions were chosen for the pilot project. For those strata underrepresented by the manual picks, a fourth region was chosen, resulting in a total of 30 regions. For all strata, a "backup" region was designated for use in the event of unforeseen technical problems.
In greater detail, the stratification criteria were as follows:
- Gene density: The gene density score of a region was the percentage of bases covered either by genes in the Ensembl database, or by human mRNA best BLAT (BLAST-like alignment tool) alignments in the UCSC Genome Browser database.
- Non-exonic conservation: The region was divided into non-overlapping subwindows of 125 bases. Subwindows that showed less than 75% base alignment with mouse sequence were discarded. For the remaining subwindows, the percentage with at least 80% base identity to mouse, and which did not correspond to Ensembl genes, GenBank mRNA BLASTZ alignments, Fgenesh++ gene predictions, TwinScan gene predictions, spliced EST alignments, or repeated sequences (DNA), was used as the non-exonic conservation score.
The above scores were computed within non-overlapping 500 kb windows of finished sequence across the genome, and used to assign each window to a stratum.[18]
Pilot Phase Results
The pilot phase was successfully finished and the results were published in June 2007 in Nature[8] and in a special issue of Genome Research;[19] the results published in the first paper mentioned advanced the collective knowledge about human genome function in several major areas, included in the following highlights:[8]
- The human genome is pervasively transcribed, such that the majority of its bases are associated with at least one primary transcript and many transcripts link distal regions to established protein-coding loci.
- Many novel non-protein-coding transcripts have been identified, with many of these overlapping protein-coding loci and others located in regions of the genome previously thought to be transcriptionally silent.
- Numerous previously unrecognized transcription start sites have been identified, many of which show chromatin structure and sequence-specific protein-binding properties similar to well-understood promoters.
- Regulatory sequences that surround transcription start sites are symmetrically distributed, with no bias towards upstream regions.
- Chromatin accessibility and histone modification patterns are highly predictive of both the presence and activity of transcription start sites.
- Distal DNaseI hypersensitive sites have characteristic histone modification patterns that reliably distinguish them from promoters; some of these distal sites show marks consistent with insulator function.
- DNA replication timing is correlated with chromatin structure.
- A total of 5% of the bases in the genome can be confidently identified as being under evolutionary constraint in mammals; for approximately 60% of these constrained bases, there is evidence of function on the basis of the results of the experimental assays performed to date.
- Although there is general overlap between genomic regions identified as functional by experimental assays and those under evolutionary constraint, not all bases within these experimentally defined regions show evidence of constraint.
- Different functional elements vary greatly in their sequence variability across the human population and in their likelihood of residing within a structurally variable region of the genome.
- Surprisingly, many functional elements are seemingly unconstrained across mammalian evolution. This suggests the possibility of a large pool of neutral elements that are biochemically active but provide no specific benefit to the organism. This pool may serve as a 'warehouse' for natural selection, potentially acting as the source of lineage-specific elements and functionally conserved but non-orthologous elements between species.
The ENCODE Phase II Project: The Production Phase Project

In September 2007, National Human Genome Research Institute (NHGRI) began funding the production phase of the ENCODE project. In this phase, the goal was to analyze the entire genome and to conduct "additional pilot-scale studies".[20]
As in the pilot project, the production effort is organized as an open consortium. In October 2007, NHGRI awarded grants totaling more than $80 million over four years.[21] The production phase also includes a Data Coordination Center, a Data Analysis Center, and a Technology Development Effort.[22] At that time the project evolved into a truly global enterprise, involving 440 scientists from 32 laboratories worldwide. Once the pilot phase was completed, the project "scaled up" in 2007, profiting immensely from new-generation sequencing machines. And the data was, indeed, big; researchers generated around 15 terabytes of raw data.
By 2010, over 1,000 genome-wide data sets had been produced by the ENCODE project. Taken together, these data sets show which regions are transcribed into RNA, which regions are likely to control the genes that are used in a particular type of cell, and which regions are associated with a wide variety of proteins. The primary assays used in ENCODE are ChIP-seq, DNase I Hypersensitivity, RNA-seq, and assays of DNA methylation.
Production Phase Results
In September 2012, the project released a much more extensive set of results, in 30 papers published simultaneously in several journals, including six in Nature, six in Genome Biology and a special issue with 18 publications of Genome Research.[23]
The authors described the production and the initial analysis of 1,640 data sets designed to annotate functional elements in the entire human genome, integrating results from diverse experiments within cell types, related experiments involving 147 different cell types, and all ENCODE data with other resources, such as candidate regions from genome-wide association studies (GWAS) and evolutionary constrained regions. Together, these efforts revealed important features about the organization and function of the human genome, which were summarized in an overview paper as follows:[24]
- The vast majority (80.4%) of the human genome participates in at least one biochemical RNA and/or chromatin associated event in at least one cell type. Much of the genome lies close to a regulatory event: 95% of the genome lies within 8kb of a DNA-protein interaction (as assayed by bound ChIP-seq motifs or DNaseI footprints), and 99% is within 1.7kb of at least one of the biochemical events measured by ENCODE.
- Primate-specific elements as well as elements without detectable mammalian constraint show, in aggregate, evidence of negative selection; thus some of them are expected to be functional.
- Classifying the genome into seven chromatin states suggests an initial set of 399,124 regions with enhancer-like features and 70,292 regions with promoters-like features, as well hundreds of thousands of quiescent regions. High-resolution analyses further subdivide the genome into thousands of narrow states with distinct functional properties.
- It is possible to quantitatively correlate RNA sequence production and processing with both chromatin marks and transcription factor (TF) binding at promoters, indicating that promoter functionality can explain the majority of RNA expression variation.
- Many non-coding variants in individual genome sequences lie in ENCODE- annotated functional regions; this number is at least as large as those that lie in protein coding genes.
- SNPs associated with disease by GWAS are enriched within non-coding functional elements, with a majority residing in or near ENCODE-defined regions that are outside of protein coding genes. In many cases, the disease phenotypes can be associated with a specific cell type or TF.
The most striking finding was that the fraction of human DNA that is biologically active is considerably higher than even the most optimistic previous estimates. In an overview paper, the ENCODE Consortium reported that its members were able to assign biochemical functions to over 80% of the genome.[24] Much of this was found to be involved in controlling the expression levels of coding DNA, which makes up less than 1% of the genome.
The most important new elements of the "encyclopedia" include:
- A comprehensive map of DNase 1 hypersensitive sites, which are markers for regulatory DNA that is typically located adjacent to genes and allows chemical factors to influence their expression. The map identified nearly 3 million sites of this type, including nearly all that were previously known and many that are novel.[25]
- A lexicon of short DNA sequences that form recognition motifs for DNA-binding proteins. Approximately 8.4 million such sequences were found, comprising a fraction of the total DNA roughly twice the size of the exome. Thousands of transcription promoters were found to make use of a single stereotyped 50-base-pair footprint.[26]
- A preliminary sketch of the architecture of the network of human transcription factors, that is, factors that bind to DNA in order to promote or inhibit the expression of genes. The network was found to be quite complex, with factors that operate at different levels as well as numerous feedback loops of various types.[27]
- A measurement of the fraction of the human genome that is capable of being transcribed into RNA. This fraction was estimated to add up to more than 75% of the total DNA, a much higher value than previous estimates. The project also began to characterize the types of RNA transcripts that are generated at various locations.[28]
Data Management and Analysis
Capturing, storing, integrating, and displaying the diverse data generated is challenging. The ENCODE Data Coordination Center (DCC) organizes and displays the data generated by the labs in the consortium, and ensures that the data meets specific quality standards when it is released to the public. Before a lab submits any data, the DCC and the lab draft a data agreement that defines the experimental parameters and associated metadata. The DCC validates incoming data to ensure consistency with the agreement. It also ensures that all data is annotated using appropriate Ontologies.[29] It then loads the data onto a test server for preliminary inspection, and coordinates with the labs to organize the data into a consistent set of tracks. When the tracks are ready, the DCC Quality Assurance team performs a series of integrity checks, verifies that the data is presented in a manner consistent with other browser data, and perhaps most importantly, verifies that the metadata and accompanying descriptive text are presented in a way that is useful to our users. The data is released on the public UCSC Genome Browser website only after all of these checks have been satisfied. In parallel, data is analyzed by the ENCODE Data Analysis Center, a consortium of analysis teams from the various production labs plus other researchers. These teams develop standardized protocols to analyze data from novel assays, determine best practices, and produce a consistent set of analytic methods such as standardized peak callers and signal generation from alignment pile-ups.[30]
The National Human Genome Research Institute (NHGRI) has identified ENCODE as a "community resource project". This important concept was defined at an international meeting held in Ft. Lauderdale in January 2003 as a research project specifically devised and implemented to create a set of data, reagents, or other material whose primary utility will be as a resource for the broad scientific community. Accordingly, the ENCODE data release policy stipulates that data, once verified, will be deposited into public databases and made available for all to use without restriction.[30]
Other Projects
With the continuation of the third phase, the ENCODE Consortium has become involved with additional projects whose goals run parallel to the ENCODE project. Some of these projects were part of the second phase of ENCODE.
modENCODE project
The MODel organism ENCyclopedia Of DNA Elements (modENCODE) project is a continuation of the original ENCODE project targeting the identification of functional elements in selected model organism genomes, specifically Drosophila melanogaster and Caenorhabditis elegans.[31] The extension to model organisms permits biological validation of the computational and experimental findings of the ENCODE project, something that is difficult or impossible to do in humans.[31] Funding for the modENCODE project was announced by the National Institutes of Health (NIH) in 2007 and included several different research institutions in the US.[32][33] The project completed its work in 2012.
In late 2010, the modENCODE consortium unveiled its first set of results with publications on annotation and integrative analysis of the worm and fly genomes in Science.[34][35] Data from these publications is available from the modENCODE web site.[36]
modENCODE was run as a Research Network and the consortium was formed by 11 primary projects, divided between worm and fly. The projects spanned the following:
- Gene structure
- mRNA and ncRNA expression profiling
- Transcription factor binding sites
- Histone modifications and replacement
- Chromatin structure
- DNA replication initiation and timing
- Copy number variation.[37]
modERN
modERN, short for the model organism encyclopedia of regulatory networks, branched from the modENCODE project. The project has merged the C. elegans and Drosophila groups and focuses on the identification of additional transcription factor binding sites of the respective organisms. The project began at the same time as Phase III of ENCODE, and plans to end in 2017.[38] To date, the project has released 198 experiments,[39] with around 500 other experiments submitted and currently being processed by the DCC.
Genomics of Gene Regulation
In early 2015, the NIH launched the Genomics of Gene Regulation (GGR) program.[40] The goal of the program, which will last for three years, is to study gene networks and pathways in different systems of the body, with the hopes to further understand the mechanisms controlling gene expressions. Although the ENCODE project is separate from GGR, the ENCODE DCC has been hosting GGR data in the ENCODE portal.[41]
Roadmap
In 2008, NIH began the Roadmap Epigenomics Mapping Consortium, whose goal was to produce "a public resource of human epigenomic data to catalyze basic biology and disease-oriented research".[42] In February 2015, the consortium released an article titled "Integrative analysis of 111 reference human epigenomes" that fulfilled the consortium's goal. The consortium integrated information and annotated regulatory elements across 127 reference epigenomes, 16 of which were part of the ENCODE project.[43] Data for the Roadmap project can either be found in the Roadmap portal or ENCODE portal.
fruitENCODE project
The fruitENCODE: an encyclopedia of DNA elements for fruit ripening is a plant ENCODE project that aims to generate DNA methylation, histone modifications, DHS, gene expression, transcription factor binding datasets for all fleshy fruit species at different developmental stages. Prerelease data can be found in the fruitENCODE portal.
Criticism of the project
Although the consortium claims they are far from finished with the ENCODE project, many reactions to the published papers and the news coverage that accompanied the release were favorable. The Nature editors and ENCODE authors "... collaborated over many months to make the biggest splash possible and capture the attention of not only the research community but also of the public at large".[45] The ENCODE project's claim that 80% of the human genome has biochemical function[24] was rapidly picked up by the popular press who described the results of the project as leading to the death of junk DNA.[46][47]
However the conclusion that most of the genome is "functional" has been criticized on the grounds that ENCODE project used a liberal definition of "functional", namely anything that is transcribed must be functional. This conclusion was arrived at despite the widely accepted view, based on genomic conservation estimates from comparative genomics, that many DNA elements such as pseudogenes that are transcribed are nevertheless non-functional. Furthermore, the ENCODE project has emphasized sensitivity over specificity leading possibly to the detection of many false positives.[48][49][50] Somewhat arbitrary choice of cell lines and transcription factors as well as lack of appropriate control experiments were additional major criticisms of ENCODE as random DNA mimics ENCODE-like 'functional' behavior.[51]
In response to some of the criticisms, other scientists argued that the wide spread transcription and splicing that is observed in the human genome directly by biochemical testing is a more accurate indicator of genetic function than genomic conservation estimates because conservation estimates are all relative and difficult to align due to incredible variations in genome sizes of even closely related species, it is partially tautological, and these estimates are not based on direct testing for functionality on the genome.[52][53] Conservation estimates may be used to provide clues to identify possible functional elements in the genome, but it does not limit or cap the total amount of functional elements that could possibly exist in the genome.[53] Furthermore, much of the genome that is being disputed by critics seems to be involved in epigenetic regulation such as gene expression and appears to be necessary for the development of complex organisms.[52][54] The ENCODE results were not necessarily unexpected since increases in attributions of functionality were foreshadowed by previous decades of research.[52][54] Additionally, others have noted that the ENCODE project from the very beginning had a scope that was based on seeking biomedically relevant functional elements in the genome not evolutionary functional elements, which are not necessarily the same thing since evolutionary selection is neither sufficient nor necessary to establish a function. It is a very useful proxy to relevant functions, but an imperfect one and not the only one.[55] The ENCODE project noted that its data is used to illuminate biological and biomedical questions on genomes and function.[56]
In response to the complaints about the definition of the word "function" some have noted that ENCODE did define what it meant and since the scope of ENCODE was seeking biomedically relevant functional elements in the genome, then the conclusion of the project should be interpreted "as saying that 80 % of the genome is engaging in relevant biochemical activities that are very likely to have causal roles in phenomena deemed relevant to biomedical research." [55] Ewan Birney, one of the ENCODE researchers, commented that "function" was used pragmatically to mean "specific biochemical activity" which included different classes of assays: RNA, "broad" histone modifications, "narrow" histone modifications, DNaseI hypersensitive sites, Transcription Factor ChIP-seq peaks, DNaseI Footprints, Transcription Factor bound motifs, and Exons.[57]
In 2014, ENCODE researchers noted that in the literature, functional parts of the genome have been identified differently in previous studies depending on the approaches used. There have been three general approaches used to identify functional parts of the human genome: genetic approaches (which rely on changes in phenotype), evolutionary approaches (which rely on conservation) and biochemical approaches (which rely on biochemical testing and was used by ENCODE). All three have limitations: genetic approaches may miss functional elements that do not manifest physically on the organism, evolutionary approaches have difficulties using accurate multispecies sequence alignments since genomes of even closely related species vary considerably, and with biochemical approaches, though having high reproducibility, the biochemical signatures do not always automatically signify a function. They concluded that in contrast to evolutionary and genetic evidence, biochemical data offer clues about both the molecular function served by underlying DNA elements and the cell types in which they act and ultimately all three approaches can be used in a complementary way to identify regions that may be functional in human biology and disease. Furthermore, they noted that the biochemical maps provided by ENCODE were the most valuable things from the project since they provide a starting point for testing how these signatures relate to molecular, cellular, and organismal function.[53]
The project has also been criticized for its high cost (~$400 million in total) and favoring big science which takes money away from highly productive investigator-initiated research.[58] The pilot ENCODE project cost an estimated $55 million; the scale-up was about $130 million and the US National Human Genome Research Institute NHGRI could award up to $123 million for the next phase. Some researchers argue that a solid return on that investment has yet to be seen. There have been attempts to scour the literature for the papers in which ENCODE plays a significant part and since 2012 there have been 300 papers, 110 of which come from labs without ENCODE funding. An additional problem is that ENCODE is not a unique name dedicated to the ENCODE project exclusively, so the word 'encode' comes up in many genetics and genomics literature.[59]
Another major critique is that the results do not justify the amount of time spent on the project and that the project itself is essentially unfinishable. Although often compared to Human Genome Project (HGP) and even termed as the HGP next step, the HGP had a clear endpoint which ENCODE currently lacks.
The authors seem to sympathize with the scientific concerns and at the same time try to justify their efforts by giving interviews and explaining ENCODE details not just to the scientific public, but also to mass media. They also claim that it took more than half a century from the realization that DNA is the hereditary material of life to the human genome sequence, so that their plan for the next century would be to really understand the sequence itself.[59]
FactorBook
The analysis of transcription factor binding data generated by the ENCODE project is currently available in the web-accessible repository FactorBook.[60] Essentially, Factorbook.org is a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. In the first release, Factorbook contains:
- 457 ChIP-seq datasets on 119 TFs in a number of human cell lines
- The average profiles of histone modifications and nucleosome positioning around the TF-binding regions
- Sequence motifs enriched in the regions and the distance and orientation preferences between motif sites.[61]
See also
References
- Hong EL, Sloan CA, Chan ET, Davidson JM, Malladi VS, Strattan JS, Hitz BC, Gabdank I, Narayanan AK, Ho M, Lee BT, Rowe LD, Dreszer TR, Roe GR, Podduturi NR, Tanaka F, Hilton JA, Cherry JM (January 2016). "Principles of metadata organization at the ENCODE data coordination center. (2016 update)". Database. 2016: baw001. doi:10.1093/database/baw001. PMC 4792520. PMID 26980513.
- "The ENCODE Project: Project Overview". www.endodeproject.org. Retrieved 2023-02-23.
- "Data Use, Software, and Analysis Release Policies – ENCODE". www.encodeproject.org. Retrieved 2021-12-18.
- "The ENCODE Project: Project Overview". www.endodeproject.org. Retrieved 2023-02-23.
- Raney BJ, Cline MS, Rosenbloom KR, Dreszer TR, Learned K, Barber GP, Meyer LR, Sloan CA, Malladi VS, Roskin KM, Suh BB, Hinrichs AS, Clawson H, Zweig AS, Kirkup V, Fujita PA, Rhead B, Smith KE, Pohl A, Kuhn RM, Karolchik D, Haussler D, Kent WJ (January 2011). "ENCODE whole-genome data in the UCSC genome browser (2011 update)". Nucleic Acids Res. 39 (Database issue): D871–5. doi:10.1093/nar/gkq1017. PMC 3013645. PMID 21037257.
- The ENCODE Project Consortium (2004). "The ENCODE (ENCyclopedia Of DNA Elements) Project". Science. 306 (5696): 636–640. Bibcode:2004Sci...306..636E. doi:10.1126/science.1105136. PMID 15499007. S2CID 22837649.
- ENCODE Project Consortium (2011). Becker PB (ed.). "A User's Guide to the Encyclopedia of DNA Elements (ENCODE)". PLOS Biology. 9 (4): e1001046. doi:10.1371/journal.pbio.1001046. PMC 3079585. PMID 21526222.

- ENCODE Project Consortium, Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, et al. (2007). "Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project". Nature. 447 (7146): 799–816. Bibcode:2007Natur.447..799B. doi:10.1038/nature05874. PMC 2212820. PMID 17571346.
- Guigó R, Flicek P, Abril JF, Reymond A, Lagarde J, Denoeud F, Antonarakis S, Ashburner M, Bajic VB, Birney E, Castelo R, Eyras E, Ucla C, Gingeras TR, Harrow J, Hubbard T, Lewis SE, Reese MG (2006). "EGASP: The human ENCODE Genome Annotation Assessment Project". Genome Biology. 7 (Suppl 1): S2.1–31. doi:10.1186/gb-2006-7-s1-s2. PMC 1810551. PMID 16925836.
- "The ENCODE Project: Project Overview". www.endodeproject.org. Retrieved 2023-02-23.
- Davis, Carrie A.; Hitz, Benjamin C.; Sloan, Cricket A.; Chan, Esther T.; Davidson, Jean M.; Gabdank, Idan; Hilton, Jason A.; Jain, Kriti; Baymuradov, Ulugbek K.; Narayanan, Aditi K.; Onate, Kathrina C. (2018-01-04). "The Encyclopedia of DNA elements (ENCODE): data portal update". Nucleic Acids Research. 46 (D1): D794–D801. doi:10.1093/nar/gkx1081. ISSN 1362-4962. PMC 5753278. PMID 29126249.
- "The ENCODE Project: ENCyclopedia Of DNA Elements". www.genome.gov. Retrieved 2016-05-13.
- Saey, Tina Hesman (6 October 2012). "Team releases sequel to the human genome". Society for Science & the Public. Retrieved 18 October 2012.
- "Fig. 3: Publications using ENCODE data. | Nature". Natureevents Directory. ISSN 1476-4687.
- GmbH, Eurice. "United States of America · IHEC". ihec-epigenomes.org. Retrieved 2017-07-18.
- "ENCODE Project". www.genome.gov. Archived from the original on 2016-05-17. Retrieved 2016-05-16.
- ENCODE Program Staff (2012-10-18). "ENCODE: Pilot Project: overview". National Human Genome Research Institute.
- ENCODE Program Staff (2012-02-19). "ENCODE: Pilot Project: Target Selection". National Human Genome Research Institute.
- Weinstock GM (2007). "ENCODE: More genomic empowerment". Genome Research. 17 (6): 667–668. doi:10.1101/gr.6534207. PMID 17567987.
- "Genome.gov | ENCODE and modENCODE Projects". The ENCODE Project: ENCyclopedia Of DNA Elements. United States National Human Genome Research Institute. 2011-08-01. Retrieved 2011-08-05.
- "National Human Genome Research Institute - Organization". The NIH Almanac. United States National Institutes of Health. Retrieved 2011-08-05.
- "Genome.gov | ENCODE Participants and Projects". The ENCODE Project: ENCyclopedia Of DNA Elements. United States National Human Genome Research Institute. 2011-08-01. Retrieved 2011-08-05.
- Ecker JR, Bickmore WA, Barroso I, Pritchard JK, Gilad Y, Segal E (September 2012). "Genomics: ENCODE explained". Nature. 489 (7414): 52–5. Bibcode:2012Natur.489...52E. doi:10.1038/489052a. PMID 22955614. S2CID 5366257.
- Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M (September 2012). "An integrated encyclopedia of DNA elements in the human genome". Nature. 489 (7414): 57–74. Bibcode:2012Natur.489...57T. doi:10.1038/nature11247. PMC 3439153. PMID 22955616.
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, et al. (September 2012). "The accessible chromatin landscape of the human genome". Nature. 489 (7414): 75–82. Bibcode:2012Natur.489...75T. doi:10.1038/nature11232. PMC 3721348. PMID 22955617.
- Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, Thurman RE, John S, Sandstrom R, et al. (September 2012). "An expansive human regulatory lexicon encoded in transcription factor footprints". Nature. 489 (7414): 83–90. Bibcode:2012Natur.489...83N. doi:10.1038/nature11212. PMC 3736582. PMID 22955618.
- Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, et al. (September 2012). "Architecture of the human regulatory network derived from ENCODE data". Nature. 489 (7414): 91–100. Bibcode:2012Natur.489...91G. doi:10.1038/nature11245. PMC 4154057. PMID 22955619.
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, et al. (September 2012). "Landscape of transcription in human cells". Nature. 489 (7414): 101–8. Bibcode:2012Natur.489..101D. doi:10.1038/nature11233. PMC 3684276. PMID 22955620.
- Malladi VS, Erickson DT, Podduturi NR, Rowe LD, Chan ET, Davidson JM, Hitz BC, Ho M, Lee BT, Miyasato S, Roe GR, Simison M, Sloan CA, Strattan JS, Tanaka F, Kent WJ, Cherry JM, Hong EL (2015). "Ontology application and use at the ENCODE DCC". Database (Oxford). 2015. doi:10.1093/database/bav010. PMC 4360730. PMID 25776021.
- Brian J. Raney; et al. (2010-10-30). "ENCODE whole-genome data in the UCSC genome browser (2011 update)". Nucleic Acids Res. Nucleic Acids Research. 39 (Database issue): D871–5. doi:10.1093/nar/gkq1017. PMC 3013645. PMID 21037257.
- "The modENCODE Project: Model Organism ENCyclopedia Of DNA Elements (modENCODE)". NHGRI website. Retrieved 2008-11-13.
- "modENCODE Participants and Projects". NHGRI website. Retrieved 2008-11-13.
- "Berkeley Lab Life Sciences Awarded NIH Grants for Fruit Fly, Nematode Studies". Lawrence Berkeley National Laboratory website. 2007-05-14. Retrieved 2008-11-13.
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, et al. (2010). "Integrative Analysis of the Caenorhabditis elegans Genome by the modENCODE Project". Science. 330 (6012): 1775–1787. Bibcode:2010Sci...330.1775G. doi:10.1126/science.1196914. PMC 3142569. PMID 21177976.
- modENCODE Consortium, Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L, et al. (2010). "Identification of Functional Elements and Regulatory Circuits by Drosophila modENCODE". Science. 330 (6012): 1787–1797. Bibcode:2010Sci...330.1787R. doi:10.1126/science.1198374. PMC 3192495. PMID 21177974.
- "modENCODE". The National Human Genome Research Institute.
- Celniker S (2009-06-11). "Unlocking the secrets of the genome". Nature. 459 (7249): 927–930. Bibcode:2009Natur.459..927C. doi:10.1038/459927a. PMC 2843545. PMID 19536255.
- "RePORT ⟩ RePORTER".
- "Search – ENCODE".
- "2015 Release: NIH grants aim to decipher the language of gene regulation". www.genome.gov. Archived from the original on 2016-04-06.
- "Search – ENCODE".
- "Roadmap Epigenomics Project - Home".
- Kundaje, Anshul; Meuleman, Wouter; Ernst, Jason; Bilenky, Misha; Yen, Angela; Heravi-Moussavi, Alireza; Kheradpour, Pouya; Zhang, Zhizhuo; Wang, Jianrong; Ziller, Michael J.; Amin, Viren; Whitaker, John W.; Schultz, Matthew D.; Ward, Lucas D.; Sarkar, Abhishek; Quon, Gerald; Sandstrom, Richard S.; Eaton, Matthew L.; Wu, Yi-Chieh; Pfenning, Andreas R.; Wang, Xinchen; Claussnitzer, Melina; Liu, Yaping; Coarfa, Cristian; Harris, R. Alan; Shoresh, Noam; Epstein, Charles B.; Gjoneska, Elizabeta; Leung, Danny; et al. (2015). "Integrative analysis of 111 reference human epigenomes". Nature. 518 (7539): 317–330. Bibcode:2015Natur.518..317.. doi:10.1038/nature14248. PMC 4530010. PMID 25693563.
- Cho, Young-Dan; Kim, Woo-Jin; Ryoo, Hyun-Mo; Kim, Hong-Gee; Kim, Kyoung-Hwa; Ku, Young; Seol, Yang-Jo (2021-04-26). "Current advances of epigenetics in periodontology from ENCODE project: a review and future perspectives". Clinical Epigenetics. 13 (1): 92. doi:10.1186/s13148-021-01074-w. ISSN 1868-7083. PMC 8077755. PMID 33902683. S2CID 233402899.
- Maher B (2012-09-06). "Fighting about ENCODE and junk". News Blog. Nature Publishing Group.
- Kolata G (2012-09-05). "Far From 'Junk,' DNA Dark Matter Proves Crucial to Health". The New York Times.
- Gregory TR (2012-09-06). "The ENCODE media hype machine". Genomicron.
- Graur D, Zheng Y, Price N, Azevedo RB, Zufall RA, Elhaik E (2013). "On the immortality of television sets: "function" in the human genome according to the evolution-free gospel of ENCODE". Genome Biol Evol. 5 (3): 578–90. doi:10.1093/gbe/evt028. PMC 3622293. PMID 23431001.
- Moran LA (2013-03-15). "Sandwalk: On the Meaning of the Word "Function"". Sandwalk.
- Gregory TR (2013-04-11). "Critiques of ENCODE in peer-reviewed journals. « Genomicron". Genomicron. Archived from the original on 21 April 2013.
- White MA, Myers CA, Corbo JC, Cohen BA (July 2013). "Massively parallel in vivo enhancer assay reveals that highly local features determine the cis-regulatory function of ChIP-seq peaks". Proc. Natl. Acad. Sci. U.S.A. 110 (29): 11952–7. Bibcode:2013PNAS..11011952W. doi:10.1073/pnas.1307449110. PMC 3718143. PMID 23818646.
- Mike White (17 July 2013). "Finding function in the genome with a null hypothesis". The Finch & Pea.
- Mattick JS, Dinger ME (2013). "The extent of functionality in the human genome". The HUGO Journal. 7 (1): 2. doi:10.1186/1877-6566-7-2. PMC 4685169.
- Kellis M, et al. (2014). "Defining functional DNA elements in the human genome". Proc. Natl. Acad. Sci. U.S.A. 111 (17): 6131–8. Bibcode:2014PNAS..111.6131K. doi:10.1073/pnas.1318948111. PMC 4035993. PMID 24753594.
- Carey, Nessa (2015). Junk DNA: A Journey Through the Dark Matter of the Genome. Columbia University Press. ISBN 9780231170840.
- Germain, Pierre-Luc; Ratti, Emanuele; Boem, Federico (November 2014). "Junk or Functional DNA? ENCODE and the Function Controversy". Biology & Philosophy. 29 (6): 807–831. doi:10.1007/s10539-014-9441-3. S2CID 84480632.
- Abascal F, Acosta R, Addleman NJ, Adrian J, et al. (30 July 2020). "Expanded Encyclopaedias of DNA elements in the Human and Mouse Genomes". Nature. 583 (7818): 699–710. Bibcode:2020Natur.583..699E. doi:10.1038/s41586-020-2493-4. PMC 7410828. PMID 32728249.
An accompanying Perspective provides further context for the evolution of the ENCODE Project and describes how ENCODE data are being used to illuminate both basic biological and biomedical questions that intersect genome structure and function.
- Birney, Ewan (September 5, 2012). "ENCODE: My own thoughts". Ewan's Blog: Bioinformatician at large.
- Timpson T (2013-03-05). "Debating ENCODE: Dan Graur, Michael Eisen". Mendelspod.
- Maher B (September 2012). "ENCODE: The human encyclopaedia". Nature. 489 (7414): 46–8. doi:10.1038/489046a. PMID 22962707.
- FactorBook
- Wang J (2012-11-29). "Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium". Nucleic Acids Research. 41 (Database issue): D171-6. doi:10.1093/nar/gks1221. PMC 3531197. PMID 23203885.
External links
- Official website
- Official list of ENCODE project publications
- ENCODE project at the National Human Genome Research Institute
- Encyclopedia of DNA Elements at the UCSC Genome Browser
- ENCODE/GENCODE project at the Wellcome Trust Sanger Institute
- ENCODE-sponsored introductory tutorial
- FactorBook
- modENCODE
- ENCODE threads Explorer at the Nature (journal)