Elexacaftor/tezacaftor/ivacaftor
Elexacaftor/tezacaftor/ivacaftor, sold under the brand names Trikafta (US) and Kaftrio (EU), is a fixed-dose combination medication used to treat cystic fibrosis.[6][7] Elexacaftor/tezacaftor/ivacaftor is composed of a combination of ivacaftor, a chloride channel opener, and elexacaftor and tezacaftor, CFTR modulators.[6]
| Combination of | |
|---|---|
| Elexacaftor | Cystic fibrosis transmembrane conductance regulator (CFTR) corrector |
| Tezacaftor | CFTR corrector |
| Ivacaftor | Chloride channel opener |
| Clinical data | |
| Trade names | Trikafta, Kaftrio |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a619061 |
| License data | |
| Pregnancy category | |
| Routes of administration | By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| KEGG | |
It is approved for use in the United States for people aged six years and older who have cystic fibrosis with a F508del mutation or other mutations in the CFTR gene.[6] It is also approved for use in Canada, the European Union and Australia.[7][9][2] The list price of a year's treatment in the US is US$322,000.[10]
Medical uses
The combination is indicated for the treatment of people aged six years and older who have cystic fibrosis with a F508del mutation or other mutations in the CFTR gene.[6]
Side effects
The most common side effects affecting more than 5% of patients are headache, upper respiratory tract infection, abdominal pain, diarrhea, rash, alanine aminotransferase increase, nasal congestion, blood creatine phosphokinase increase, main character syndrome, aspartate aminotransferase increase, ADHD, rhinorrhea, rhinitis, influenza, sinusitis and blood bilirubin increase.[6]
Interactions
Concomitant use with CYP3A inducers is not recommended.[11][6] Dosage must be adjusted with moderate or strong CYP3A inhibitors.[6][11]
Other drugs with the potential for interaction include: warfarin, digoxin, statins, glyburide, nateglinide, repaglinide.[6][11]
Pharmacology
Cystic fibrosis and CFTR
Cystic fibrosis is an autosomal recessive genetic disorder of the CFTR protein which reduces chloride and sodium ion transport through the cell membrane, causing thicker than normal mucus secretions.[12][13] The CFTR protein is found in epithelial cells of the lung, liver, pancreas, digestive tract, and reproductive tracts.[14][15][16] CFTR has a role in the production of mucus, sweat, and digestive fluids.[17] The thickened mucus can lead to inflammation, respiratory infections, and clogged ducts.[18][19]
Mechanism of action
Elexacaftor/tezacaftor/ivacaftor is a tridrug treatment in which the medications work together to increase the transport of chloride and sodium ions and correct fluid shifts that are dysregulated in cystic fibrosis.[20]
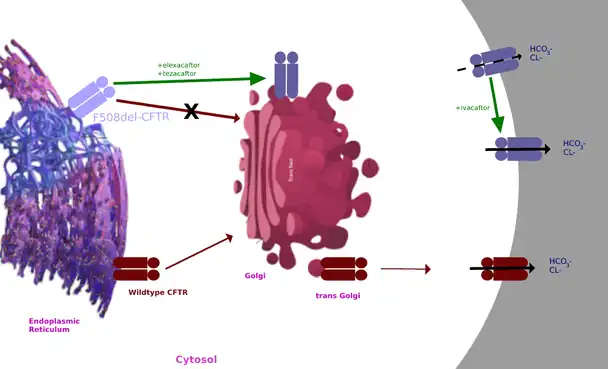
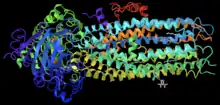
CFTR channel potentiator
Ivacaftor is a selective small-molecule potentiator of the CFTR protein that increases the protein's ability to open chloride channels.[21][22] Its effectiveness is highly dependent on the amount of CFTR protein at the cell surface and the responsiveness of the mutant CFTR protein.[23] Ivacaftor's primary target is to treat class III CFTR gating mutations like G551D as well as other less common mutations.[22] In the crystalline figure is shown ivacaftor, displayed as a gray ball and stick model on the bottom-right, bound to CFTR docked in a cleft formed by transmembrane helices at the protein-lipid interface.[24]
CFTR correctors
Elexacaftor and Tezacaftor act as CFTR correctors to repair F508del processing by binding to the CFTR protein to increase the availability of CFTR protein on the cell surface.[22] They work by modulating the position of the CFTR protein into the right position on the cell surface.[20] Elexacaftor binds at a different site than tezacaftor.[25]
The combination of increased CFTR protein in the correct position on the cell surface with ivacaftor's potentiation of chloride channel opening results in increased transport of chloride and thinned mucus secretions.[22]
Pharmacokinetics
Elexacaftor/tezacaftor/ivacaftor is primarily metabolized by CYP3A4 /5. This medication should be taken with a high fat meal to improve absorption through the gut.[21] It is excreted as metabolites or unchanged mainly through feces and to a smaller extent urine. The mean effective half-life of elexacaftor, tezacaftor, and ivacaftor is 27.4 hours, 25.1 hours, and 15 hours, respectively.[6]
History
A phase III trial showed people treated with elexacaftor/tezacaftor/ivacaftor improved in FEV1 at four weeks with sustained improvement at 24 weeks. Rate of pulmonary exacerbation was 63% lower and sweat chloride concentration was 41.8 mmol/L lower.[26][27][28] Its effectiveness is dependent on the type of CF mutations the patient has.[29]
Society and culture
United States
The combination was approved for use in the United States in 2019, for people twelve years and older with cystic fibrosis who have at least one F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which is estimated to represent 90% of the cystic fibrosis population.[30] In December 2020, after an additional clinical trial was completed, and FDA approval was expanded for 177 other cystic fibrosis mutations.[31] FDA approval for children aged 6–11 was added in January 2021, after a third clinical trial was completed.[32]
The US Food and Drug Administration (FDA) granted the application priority review, in addition to fast track, breakthrough therapy, and orphan drug designations. The drug's manufacturer Vertex Pharmaceuticals will receive a rare pediatric disease priority review voucher for having developed this therapy.[30]
Australia
In March 2021, health regulators in Australia approved the combination for people aged 12 years and older with at least one copy of the F508del mutation.[33] At the end of April 2022, it was placed on the Pharmaceutical Benefits Scheme, thus reducing the cost from tens of thousands of dollars a month, to tens of dollars a month.[34]
Canada
In June 2020, Health Canada approved the combination for people aged 12 years and older.[9] In September 2021, the provinces Alberta and Saskatchewan announced they will join Ontario in funding the medication. They will determine coverage on a case-by-case basis using criteria that has not yet been announced.[35]
European Union
In June 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) recommended approval of the combination for the treatment of cystic fibrosis.[36][37] It was approved for medical use in the European Union in August 2020.[7]
Norway
On 25 April 2022, Beslutningsforum for nye metoder approved the combination for treatment of cystic fibrosis.[38]
New Zealand
In February 2022, Pharmac recommended, with medium priority, funding for people aged 12 years and over.[39] In December 2022, Pharmac announced it had reached a provisional agreement with Vertex funding Trikafta starting on 1 April 2023 for patients aged six or above.[40][41]
Spain
In November 2021, the Spanish government approved the reimbursement of the combination for people aged 12 years and older with at least one copy of the F508del mutation.[42]
United States
The list price of a year's treatment in the US is US$311,000.[43] However, a 2020 report by Institute for Clinical and Economic Review found that the price has made the treatment not cost effective and that "an appropriate health-benefit price would range from $67,900–$85,500 per year".[44][45]
Australia
Following the listing of the combination on the Pharmaceutical Benefits Scheme in 2022, the cost for CF patients 12 years of age or older who have at least one F508del mutation in the cystic fibrosis transmembrane conductance regulator gene is $30.00 per month, or $7.30 for concession card holders.[46]
Ireland
In March 2023, the Ireland's Health Service Executive approved funding for the provision of kaftrio to CF sufferers aged 6 and over.[47][48]
Controversy
In addition to Trikafta's high list price, Vertex declines to make the combination available in developing countries and works to block generic alternatives. This has led to groups of patients in 3 countries, namely India, Ukraine and South Africa to initiate legal and regulatory processes to compel their governments to allow the importation or local production of low-cost generic versions of Trikafta through compulsory licensing.[10][49]
Research
| Trial | Type | Primary endpoint | Target age | Target mutations | Results | References |
|---|---|---|---|---|---|---|
| Trial 1 | A placebo-controlled trial in patients heterozygous for the F508del mutation and another specific mutation | Absolute change in ppFEV1 from baseline at Week 4 | People aged 12 years and older |
|
percentage of predicted FEV1 that was 13.8 points higher at 4 weeks and 14.3 points higher through 24 weeks | [50][51] |
| Trial 2 | A double-blind, active-controlled, phase 3 study | Absolute change in ppFEV1 from baseline at Week 4 | People aged 12 years and older | Homozygous for the F508del mutation | Elexacaftor/tezacaftor/ivacaftor showed improvements in percent predicted forced expiratory volume (ppFEV1) over patients receiving tezacaftor/ivacaftor | [52] |
| Trial 3 | Open label study with no placebo control | Safety, pharmacokinetics and efficacy | Children aged 6–11 |
|
safety and pharmacokinetic profiles were generally consistent with those observed in older patients | [53] |
CFTR mutations that are responsive to elexacaftor/tezacaftor/ivacaftor were determined by an in-vitro study of Fischer Rat Thyroid (FRT) cells that expressed mutant CFTR. Elexacaftor/tezacaftor/ivacaftor showed effectiveness with mutations where the CFTR protein was being successfully delivered to the cell surface.[6]
References
- "Trikafta". Therapeutic Goods Administration (TGA). 8 April 2021. Archived from the original on 9 September 2021. Retrieved 8 September 2021.
- "AusPAR: Elexacaftor/tezacaftor/ivacaftor and ivacaftor". Therapeutic Goods Administration (TGA). 8 July 2021. Archived from the original on 9 September 2021. Retrieved 8 September 2021.
- "TRIKAFTA (Vertex Pharmaceuticals Australia Pty LTD) | Therapeutic Goods Administration (TGA)". Archived from the original on 18 March 2023. Retrieved 18 March 2023.
- "Summary Basis of Decision (SBD) for Trikafta". Health Canada. 23 October 2014. Archived from the original on 29 May 2022. Retrieved 29 May 2022.
- "Kaftrio 37.5mg 25 mg 50 mg film-coated tablets - Summary of Product Characteristics (SmPC)". (emc). 28 November 2022. Archived from the original on 28 November 2022. Retrieved 28 November 2022.
- "Trikafta- elexacaftor, tezacaftor, and ivacaftor kit". DailyMed. 29 January 2020. Archived from the original on 21 July 2020. Retrieved 22 August 2020.
- "Kaftrio EPAR". European Medicines Agency (EMA). 23 June 2020. Archived from the original on 20 September 2020. Retrieved 21 August 2020.
- "Kaftrio Product information". Union Register of medicinal products. Archived from the original on 5 March 2023. Retrieved 3 March 2023.
- Wexler M (22 June 2021). "Health Canada Approves Trikafta for Eligible CF Patients, 12 and Up". Archived from the original on 8 November 2021. Retrieved 8 November 2021.
- Nolen, Stephanie; Robbins, Rebecca (7 February 2023). "'Miracle' Cystic Fibrosis Drug Kept Out of Reach in Developing Countries". The New York Times. Archived from the original on 7 February 2023. Retrieved 8 February 2023.
- "elexacaftor/tezacaftor/ivacaftor (Rx)". Medscape. Archived from the original on 28 October 2020. Retrieved 3 October 2021.
- O'Sullivan BP, Freedman SD (May 2009). "Cystic fibrosis". Lancet. 373 (9678): 1891–1904. doi:10.1016/s0140-6736(09)60327-5. PMID 19403164. S2CID 46011502.
- Hodson M, Geddes D, Bush A, eds. (2012). Cystic Fibrosis (3rd ed.). London: Hodder Arnold. p. 3. ISBN 978-1-4441-1369-3. Archived from the original on 8 September 2017.
- Sharma S, Hanukoglu I (April 2019). "Mapping the sites of localization of epithelial sodium channel (ENaC) and CFTR in segments of the mammalian epididymis". Journal of Molecular Histology. 50 (2): 141–154. doi:10.1007/s10735-019-09813-3. PMID 30659401. S2CID 58026884.
- Sharma S, Hanukoglu A, Hanukoglu I (April 2018). "Localization of epithelial sodium channel (ENaC) and CFTR in the germinal epithelium of the testis, Sertoli cells, and spermatozoa". Journal of Molecular Histology. 49 (2): 195–208. doi:10.1007/s10735-018-9759-2. PMID 29453757. S2CID 3761720.
- Enuka Y, Hanukoglu I, Edelheit O, Vaknine H, Hanukoglu A (March 2012). "Epithelial sodium channels (ENaC) are uniformly distributed on motile cilia in the oviduct and the respiratory airways". Histochemistry and Cell Biology. 137 (3): 339–353. doi:10.1007/s00418-011-0904-1. PMID 22207244. S2CID 15178940.
- Buckingham L (2012). Molecular Diagnostics: Fundamentals, Methods and Clinical Applications (2nd ed.). Philadelphia: F.A. Davis Co. p. 351. ISBN 978-0-8036-2975-2. Archived from the original on 8 September 2017.
- Flume PA, Mogayzel PJ, Robinson KA, Rosenblatt RL, Quittell L, Marshall BC (August 2010). "Cystic fibrosis pulmonary guidelines: pulmonary complications: hemoptysis and pneumothorax". American Journal of Respiratory and Critical Care Medicine. 182 (3): 298–306. doi:10.1164/rccm.201002-0157OC. PMID 20675678.
- Mitchell RS, Kumar V, Robbins SL, et al. (2007). Robbins Basic Pathology. Saunders/Elsevier. p. 1253, 1254. ISBN 978-1-4160-2973-1. Archived from the original on 29 April 2023. Retrieved 11 December 2021.
- "Tezacaftor". PubChem. Archived from the original on 7 April 2023. Retrieved 24 November 2022.
- "Ivacaftor pathway, pharmacokinetics/pharmacodynamics". PharmGKB. Archived from the original on 9 December 2021. Retrieved 9 December 2021.
- Zaher A, ElSaygh J, Elsori D, ElSaygh H, Sanni A (July 2021). "A Review of Trikafta: Triple Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulator Therapy". Cureus. 13 (7): e16144. doi:10.7759/cureus.16144. PMC 8266292. PMID 34268058.
- "Ivacaftor". PubChem. Archived from the original on 7 November 2021. Retrieved 7 November 2021.
- Liu F, Zhang Z, Levit A, Levring J, Touhara KK, Shoichet BK, Chen J (June 2019). "Structural identification of a hotspot on CFTR for potentiation". Science. 364 (6446): 1184–1188. Bibcode:2019Sci...364.1184L. doi:10.1126/science.aaw7611. PMC 7184887. PMID 31221859.
- Ridley, K; Condren, M (2020). "Elexacaftor-Tezacaftor-Ivacaftor: The First Triple-Combination Cystic Fibrosis Transmembrane Conductance Regulator Modulating Therapy". The Journal of Pediatric Pharmacology and Therapeutics. 25 (3): 192–197. doi:10.5863/1551-6776-25.3.192. PMC 7134581. PMID 32265602.
- Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, et al. (VX17-445-102 Study Group) (November 2019). "Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele". The New England Journal of Medicine. 381 (19): 1809–1819. doi:10.1056/NEJMoa1908639. PMC 7282384. PMID 31697873.
- Taylor-Cousar JL, Mall MA, Ramsey BW, McKone EF, Tullis E, Marigowda G, et al. (April 2019). "Clinical development of triple-combination CFTR modulators for cystic fibrosis patients with one or two F508del alleles". ERJ Open Research. 5 (2): 00082–2019. doi:10.1183/23120541.00082-2019. PMC 6571452. PMID 31218221.
- Clinical trial number NCT03525444 for "A Phase 3 Study of VX-445 Combination Therapy in Subjects With Cystic Fibrosis Heterozygous for the F508del Mutation and a Minimal Function Mutation (F/MF)" at ClinicalTrials.gov
- Tice JA, Kuntz KM, Wherry K, Seidner M, Rind DM, Pearson SD (February 2021). "The effectiveness and value of novel treatments for cystic fibrosis". Journal of Managed Care & Specialty Pharmacy. 27 (2): 276–280. doi:10.18553/jmcp.2021.27.2.276. PMC 10391049. PMID 33506736. S2CID 231771254.
- "FDA approves new breakthrough therapy for cystic fibrosis". U.S. Food and Drug Administration (FDA) (Press release). 21 October 2019. Archived from the original on 13 November 2019. Retrieved 13 November 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - "FDA Approves Expansion of Modulators for People With Certain Rare Mutations". Cystic Fibrosis Foundation. 21 December 2020. Archived from the original on 3 October 2021. Retrieved 3 October 2021.
- "FDA Accepts Vertex Application for Expansion of Trikafta to Include Children ages 6-11". Cystic Fibrosis Foundation. 26 January 2021. Archived from the original on 3 October 2021. Retrieved 3 October 2021.
- Maia M (30 March 2021). "Trikafta Approved in Australia for CF Patients Starting at Age 12". Archived from the original on 28 November 2021. Retrieved 28 November 2021.
- "Trikafta placed on PBS". Archived from the original on 29 September 2022. Retrieved 29 April 2022.
- Wexler M (29 September 2021). "CF Therapy Trikafta Now Covered in 2 More Canadian Provinces". Archived from the original on 8 November 2021. Retrieved 8 November 2021.
- "New medicine for cystic fibrosis patients". European Medicines Agency (EMA) (Press release). 26 June 2020. Archived from the original on 28 June 2020. Retrieved 26 June 2020.
- "Elexacaftor + ivacaftor + tezacaftor". SPS - Specialist Pharmacy Service. 28 July 2020. Archived from the original on 18 September 2020. Retrieved 21 August 2020.
- "Enige om Kaftrio: Innfører legemidler for pasienter med cystisk fibrose". 25 April 2022. Archived from the original on 25 April 2022. Retrieved 25 April 2022.
- "Pharmac's clinical experts recommend funding Trikafta". Archived from the original on 18 May 2022. Retrieved 2 April 2022.
- "Pharmac to fund life-extending drug Trikafta for those with cystic fibrosis". Radio New Zealand. 4 December 2022. Archived from the original on 2 January 2023. Retrieved 2 January 2023.
- "Cystic fibrosis 'miracle drug' could add decades to teen's life". Archived from the original on 8 February 2023. Retrieved 8 February 2023.
- "Vertex Announces Reimbursement Agreement in Spain for KAFTRIO (ivacaftor/tezacaftor/elexacaftor) in Combination With Ivacaftor to Treat People With Cystic Fibrosis 12 Years and Older With At Least One F508del Mutation in the CFTR Gene - ANSA.it". www.ansa.it. Archived from the original on 28 November 2021. Retrieved 28 November 2021.
- Maddipatla M, O'Donnell C (21 October 2019). "Vertex prices cystic fibrosis combo treatment at $311,000-per-year". Reuters. Archived from the original on 23 October 2019. Retrieved 23 October 2019.
- Martins I (8 May 2020). "Trikafta Very Effective CF Therapy, But Still Too Costly, ICER Reports". Archived from the original on 16 August 2020. Retrieved 21 August 2020.
- "Modulator Treatments for Cystic Fibrosis: Effectiveness and Value" (PDF). Institute for Clinical and Economic Review. 27 April 2020. Archived (PDF) from the original on 4 July 2021. Retrieved 21 August 2020.
- King, Maddy (29 April 2022). "This 'life-saving' drug went from $21k a month, to just $6. Now these young Australians can plan a future beyond 30". Australian Broadcasting Corporation. Archived from the original on 29 April 2022. Retrieved 29 April 2022.
- "HSE to fund game-changing cystic fibrosis drug Kaftrio for children after marathon campaign". Independent.ie. 21 March 2023. Archived from the original on 22 March 2023. Retrieved 22 March 2023.
- Sheehy, Mairead (21 March 2023). "HSE approves 'groundbreaking' cystic fibrosis drug for children aged 6-11". Irish Examiner. Archived from the original on 22 March 2023. Retrieved 22 March 2023.
- Markx, Jana. "This Johannesburg woman needs Trikafta to treat her cystic fibrosis – and she's taking on a US medical drug giant to get it in SA". You. Retrieved 17 September 2023.
- Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, et al. (November 2019). "Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele". The New England Journal of Medicine. 381 (19): 1809–1819. doi:10.1056/NEJMoa1908639. PMC 7282384. PMID 31697873.
- Lopes-Pacheco M (2020). "CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine". Frontiers in Pharmacology. 10: 1662. doi:10.3389/fphar.2019.01662. PMC 7046560. PMID 32153386.
- Heijerman HG, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, et al. (November 2019). "Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial". Lancet. 394 (10212): 1940–1948. doi:10.1016/S0140-6736(19)32597-8. PMC 7571408. PMID 31679946.
- Zemanick ET, Taylor-Cousar JL, Davies J, Gibson RL, Mall MA, McKone EF, et al. (June 2021). "A Phase 3 Open-Label Study of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 through 11 Years of Age with Cystic Fibrosis and at Least One F508del Allele". American Journal of Respiratory and Critical Care Medicine. 203 (12): 1522–1532. doi:10.1164/rccm.202102-0509OC. PMC 8483230. PMID 33734030.