Gene set enrichment analysis
Gene set enrichment analysis (GSEA) (also called functional enrichment analysis or pathway enrichment analysis) is a method to identify classes of genes or proteins that are over-represented in a large set of genes or proteins, and may have an association with different phenotypes (e.g. different organism growth patterns or diseases). The method uses statistical approaches to identify significantly enriched or depleted groups of genes. Transcriptomics technologies and proteomics results often identify thousands of genes which are used for the analysis.[1]
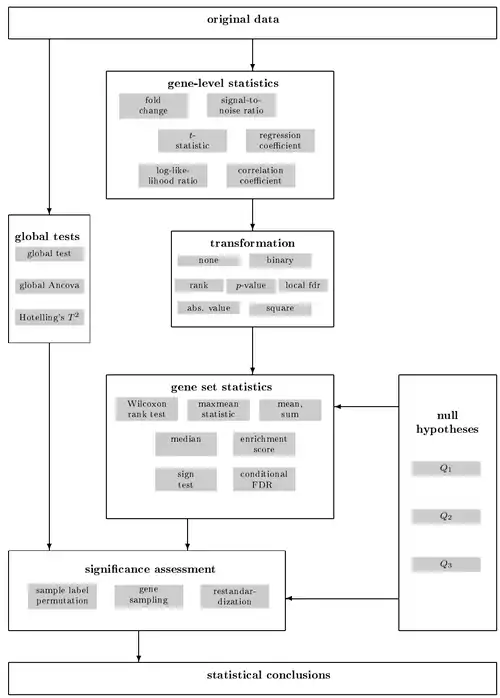
Researchers performing high-throughput experiments that yield sets of genes (for example, genes that are differentially expressed under different conditions) often want to retrieve a functional profile of that gene set, in order to better understand the underlying biological processes. This can be done by comparing the input gene set to each of the bins (terms) in the gene ontology – a statistical test can be performed for each bin to see if it is enriched for the input genes.
Background
After the completion of the Human Genome Project, the problem of how to interpret and analyze it remained. In order to seek out genes associated with diseases, DNA microarrays were used to measure the amount of gene expression in different cells. Microarrays on thousands of different genes were carried out, and comparisons the results of two different cell categories, e.g. normal cells versus cancerous cells. However, this method of comparison is not sensitive enough to detect the subtle differences between the expression of individual genes, because diseases typically involve entire groups of genes.[2] Multiple genes are linked to a single biological pathway, and so it is the additive change in expression within gene sets that leads to the difference in phenotypic expression. Gene Set Enrichment Analysis was developed [2] to focus on the changes of expression in groups of a priori defined gene sets. By doing so, this method resolves the problem of the undetectable, small changes in the expression of single genes.[3]
Methods
Gene set enrichment analysis uses a priori gene sets that have been grouped together by their involvement in the same biological pathway, or by proximal location on a chromosome.[1] A database of these predefined sets can be found at the Molecular signatures database (MSigDB).[4][5] In GSEA, DNA microarrays, or now RNA-Seq, are still performed and compared between two cell categories, but instead of focusing on individual genes in a long list, the focus is put on a gene set.[1] Researchers analyze whether the majority of genes in the set fall in the extremes of this list: the top and bottom of the list correspond to the largest differences in expression between the two cell types. If the gene set falls at either the top (over-expressed) or bottom (under-expressed), it is thought to be related to the phenotypic differences.
In the method that is typically referred to as standard GSEA, there are three steps involved in the analytical process.[1][2] The general steps are summarized below:
- Calculate the enrichment score (ES) that represents the amount to which the genes in the set are over-represented at either the top or bottom of the list. This score is a Kolmogorov–Smirnov-like statistic.[1][2]
- Estimate the statistical significance of the ES. This calculation is done by a phenotypic-based permutation test in order to produce a null distribution for the ES. The P value is determined by comparison to the null distribution.[1][2]
- Adjust for multiple hypothesis testing for when a large number of gene sets are being analyzed at one time. The enrichment scores for each set are normalized and a false discovery rate is calculated.[1][2]
This can be described as:
![{\displaystyle {\begin{alignedat}{1}&P_{hit}(S,i)=\sum _{g_{j}\in S,j\leq i}{\dfrac {|r_{j}|^{p}}{N_{R}}};\\[0.6ex]&P_{miss}(S,i)=\sum _{g_{j}\not \in S,j\leq i}{\dfrac {1}{N-N_{H}}};\\[0.6ex]&N_{R}=\sum _{g_{j}\in S}|r_{j}|^{p};\\[0.6ex]&ES=P(S,i)=P_{hit}(S,i)-P_{miss}(S,i)=max(|P_{hit}(S,i)-P_{miss}(S,i)|)\\[0.6ex]\end{alignedat}}}](../I/b57a1d21afd92a9de0451aa2dea520790353f28f.svg)
Where is the rank of the gene, is the power usually set to 1 (if it were 0, it would be equivalent to the Kolmogorov–Smirnov test).


SEA
When GSEA was first proposed in 2003 some immediate concerns were raised regarding its methodology. These criticisms led to the use of the correlation-weighted Kolmogorov–Smirnov test, the normalized ES, and the false discovery rate calculation, all of which are the factors that currently define standard GSEA.[6] However, GSEA has now also been criticized for the fact that its null distribution is superfluous, and too difficult to be worth calculating, as well as the fact that its Kolmogorov–Smirnov-like statistic is not as sensitive as the original.[6] As an alternative, the method known as Simpler Enrichment Analysis (SEA), was proposed. This method assumes gene independence and uses a simpler approach to calculate t-test. However, it is thought that these assumptions are in fact too simplifying, and gene correlation cannot be disregarded.[6]
SGSE
One other limitation to Gene Set Enrichment Analysis is that the results are very dependent on the algorithm that clusters the genes, and the number of clusters being tested.[7] Spectral Gene Set Enrichment (SGSE) is a proposed, unsupervised test. The method's founders claim that it is a better way to find associations between MSigDB gene sets and microarray data. The general steps include:
1. Calculating the association between principal components and gene sets.[7]
2. Using the weighted Z-method to calculate the association between the gene sets and the spectral structure of the data.[7]
Tools
GSEA uses complicated statistics, so it requires a computer program to run the calculations. GSEA has become standard practice, and there are many websites and downloadable programs that will provide the data sets and run the analysis.
MOET
Multi-Ontology Enrichment Tool (MOET): MOET is a web-based ontology analysis tool that provides functionality for multiple ontologies, including Disease, GO, Pathway, Phenotype, and Chemical entities (ChEBI) for multiple species, including rat, mouse, human, bonobo, squirrel, dog, pig, chinchilla, naked mole-rat and vervet (green monkey). It outputs a downloadable graph and a list of statistically overrepresented terms in the user’s list of genes using hypergeometric distribution. MOET also displays the corresponding Bonferroni correction and odds ratio on the results page. It is simple to use, and results are provided with a few clicks in seconds; no software installations or programming skills are required. In addition, MOET is updated weekly, providing the user with the most recent data for analyses.
NASQAR
NASQAR (Nucleic Acid SeQuence Analysis Resource) is an open source, web-based platform for high-throughput sequencing data analysis and visualization.[8][9] Users can perform GSEA using the popular R-based clusterProfiler package [10] in a simple, user-friendly web app. NASQAR currently supports GO Term and KEGG Pathway enrichment with all organisms supported by an Org.Db database.[11]
PlantRegMap
The gene ontology (GO) annotation for 165 plant species and GO enrichment analysis is available.[12]
MSigDB
The Molecular Signatures Database hosts an extensive collection of annotated gene sets that can be used with most GSEA Software.
Broad Institute
The Broad Institute website is in cooperation with MSigDB and has a downloadable GSEA software, as well a general tutorial for those new to performing this analytical technique.[13]
WebGestalt
WebGestalt [14] is a web based gene set analysis toolkit. It supports three well-established and complementary methods for enrichment analysis, including Over-Representation Analysis (ORA), Gene Set Enrichment Analysis (GSEA), and Network Topology-based Analysis (NTA). Analysis can be performed against 12 organisms and 321,251 functional categories using 354 gene identifiers from various databases and technology platforms.
Enrichr
Enrichr[15] [16] [17] is a gene set enrichment analysis tool for mammalian gene sets. It contains background libraries for transcription regulation, pathways and protein interactions, ontologies including GO and the human and mouse phenotype ontologies, signatures from cells treated with drugs, gene sets associated with human diseases, and expression of genes in different cells and tissues. Enrichr was developed by the Ma'ayan Laboratory at the Icahn School of Medicine at Mount Sinai.[18] The background libraries are from over 200 resources and contain over 450,000 annotated gene sets. The tool can be accessed through API and provides different ways to visualize the results.
GeneSCF
GeneSCF is a real-time based functional enrichment tool with support for multiple organisms[19] and is designed to overcome the problems associated with using outdated resources and databases.[20] Advantages of using GeneSCF: real-time analysis, users do not have to depend on enrichment tools to get updated, easy for computational biologists to integrate GeneSCF with their NGS pipeline, it supports multiple organisms, enrichment analysis for multiple gene list using multiple source database in single run, retrieve or download complete GO terms/Pathways/Functions with associated genes as simple table format in a plain text file.[21][22]
DAVID
DAVID is the database for annotation, visualization and integrated discovery, a bioinformatics tool that pools together information from most major bioinformatics sources, with the aim of analyzing large gene lists in a high-throughput manner.[23] DAVID goes beyond standard GSEA with additional functions like switching between gene and protein identifiers on the genome-wide scale,[23] however, the annotations used by DAVID was not updated since October 2016 to Dec 2021,[24] which can have a considerable impact on practical interpretation of results.[25] However, A most recent update was performed in 2021[24]
Metascape
Metascape is a biologist-oriented gene-list analysis portal.[26] Metascape integrates pathway enrichment analysis, protein complex analysis, and multi-list meta-analysis into one seamless workflow accessible through a significantly simplified user interface. Metascape maintains analysis accuracy by updating its 40 underlying knowledgebases monthly. Metascape presents results using easy-to-interpret graphics, spreadsheets, and publication quality presentations, and is freely available.[27]
AmiGO 2
The Gene Ontology (GO) consortium has also developed their own online GO term enrichment tool,[28] allowing species-specific enrichment analysis versus the complete database, coarser-grained GO slims, or custom references.[29]
GREAT
In 2010, Gill Bejerano from Stanford University released the Genomic region enrichment of annotations tool (GREAT), a software which takes advantage of regulatory domains to better associate gene ontology terms to genes.[30] Its primary purpose is to identify pathways and processes that are significantly associated with factor regulating activity. This method maps genes with regulatory regions through a hypergeometric test over genes, inferring proximal gene regulatory domains. It does this by using the total fraction of the genome associated with a given ontology term as the expected fraction of input regions associated with the term by chance. Enrichment is calculated by all regulatory regions, and several experiments were performed to validate GREAT, one of which being enrichment analyses done on 8 ChIP-seq datasets.[31]
FunRich
The Functional Enrichment Analysis (FunRich) tool[32] is mainly used for the functional enrichment and network analysis of Omics data.[33]
FuncAssociate
FuncAssociate tool enables Gene Ontology and custom enrichment analyses. It allows inputting ordered sets as well as weighted gene space files for background.[34]
InterMine
Instances of InterMine automatically provide enrichment analysis [35] for uploaded sets of genes and other biological entities.
ToppGene suite
ToppGene is a one-stop portal for gene list enrichment analysis and candidate gene prioritization based on functional annotations and protein interactions network.[36] Developed and maintained by the Division of Biomedical Informatics at Cincinnati Children's Hospital Medical Center.
QuSAGE
Quantitative Set Analysis for Gene Expression (QuSAGE) is a computational method for gene set enrichment analysis.[37] QuSAGE improves power by accounting for inter-gene correlations and quantifies gene set activity with a complete probability density function (PDF). From this PDF, P values and confidence intervals can be easily extracted. Preserving the PDF also allows for post-hoc analysis (e.g., pair-wise comparisons of gene set activity) while maintaining statistical traceability. Turner et al. extended the applicability of QuSAGE to longitudinal studies by adding functionality for general linear mixed models.[38] QuSAGE was used by the NIH/NIAID Human Immunology Project Consortium to identify baseline transcriptional signatures that were associated with human influenza vaccination responses.[39] QuSAGE is available as an R/Bioconductor package, and is maintained by the Kleinstein Lab at Yale School of Medicine.
Blast2GO
Blast2GO is a bioinformatics platform for functional annotation and analysis of genomic datasets.[40] This tool allows to perform gene set enrichment analysis (GSEA),[41] among other functions.
g:Profiler
g:Profiler is a widely used toolset for finding biological categories enriched in gene lists, conversions between gene identifiers and mappings to their orthologs. Mission of g:Profiler is to provide a reliable service based on up to date high quality data in a convenient manner across many evidence types, identifier spaces and organisms. g:Profiler relies on Ensembl as a primary data source and follows their quarterly release cycle while updating the other data sources simultaneously. g:Profiler provides modern responsive interactive web interface, standardised API, an R package gprofiler2 and libraries. The results are delivered through interactive and configurable interface. Results can be downloaded as publication ready visualisations or delimited text files. g:Profiler supports close to 500 species and strains, including vertebrates, plants, fungi, insects and parasites. By supporting user uploaded custom GMT files, g:Profiler is capable of analysing data from any organism. All past releases are maintained for reproducibility and transparency. g:Profiler is freely available for all users at https://biit.cs.ut.ee/gprofiler.
Applications
Genome-wide association studies
Single-nucleotide polymorphisms, or SNPs, are single base mutations that may be associated with diseases. One base change has the potential to affect the protein that results from that gene being expressed; however, it also has the potential to have no effect at all. Genome-wide association studies are comparisons between healthy and disease genotypes to try to find SNPs that are overrepresented in the disease genomes, and might be associated with that condition. Before GSEA, the accuracy of genome-wide SNP association studies was severely limited by a high number of false positives.[42] The theory that the SNPs contributing to a disease tend to be grouped in a set of genes that are all involved in the same biological pathway, is what the GSEA-SNP method is based on. This application of GSEA does not only aid in the discovery of disease-associated SNPs, but helps illuminate the corresponding pathways and mechanisms of the diseases.[42]
Spontaneous preterm birth
Gene set enrichment methods led to the discovery of new suspect genes and biological pathways related to spontaneous preterm births.[43] Exome sequences from women who had experienced SPTB were compared to those from females from the 1000 Genome Project, using a tool that scored possible disease-causing variants. Genes with higher scores were then run through different programs to group them into gene sets based on pathways and ontology groups. This study found that the variants were significantly clustered in sets related to several pathways, all suspects in SPTB.[43]
Cancer cell profiling
Gene set enrichment analysis can be used to understand the changes that cells undergo during carcinogenesis and metastasis. In a study, microarrays were performed on renal cell carcinoma metastases, primary renal tumors, and normal kidney tissue, and the data was analyzed using GSEA.[44] This analysis showed significant changes of expression in genes involved in pathways that have not been previously associated with the progression of renal cancer. From this study, GSEA has provided potential new targets for renal cell carcinoma therapy.
Schizophrenia
GSEA can be used to help understand the molecular mechanisms of complex disorders. Schizophrenia is a largely heritable disorder, but is also very complex, and the onset of the disease involves many genes interacting within multiple pathways, as well the interaction of those genes with environmental factors. For instance, epigenetic changes, like DNA methylation, are affected by the environment, but are also inherently dependent on the DNA itself. DNA methylation is the most well-studied epigenetic change, and was recently analyzed using GSEA in relation to schizophrenia-related intermediate phenotypes.[45] Researchers ranked genes for their correlation between methylation patterns and each of the phenotypes. They then used GSEA to look for an enrichment of genes that are predicted to be targeted by microRNAs in the progression of the disease.[45]
Depression
GSEA can help provide molecular evidence for the association of biological pathways with diseases. Previous studies have shown that long-term depression symptoms are correlated with changes in immune response and inflammatory pathways.[46] Genetic and molecular evidence was sought to support this. Researchers took blood samples from sufferers of depression, and used genome-wide expression data, along with GSEA to find expression differences in gene sets related to inflammatory pathways. This study found that those people who rated with the most severe depression symptoms also had significant expression differences in those gene sets, and this result supports the association hypothesis.[46]
See also
References
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. (October 2005). "Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles". Proceedings of the National Academy of Sciences of the United States of America. 102 (43): 15545–15550. doi:10.1073/pnas.0506580102. PMC 1239896. PMID 16199517.
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. (July 2003). "PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes". Nature Genetics. 34 (3): 267–273. doi:10.1038/ng1180. PMID 12808457. S2CID 13940856.
- Maleki F, Ovens K, Hogan DJ, Kusalik AJ (2020). "Gene Set Analysis: Challenges, Opportunities, and Future Research". Frontiers in Genetics. 11: 654. doi:10.3389/fgene.2020.00654. PMC 7339292. PMID 32695141.
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P (December 2015). "The Molecular Signatures Database (MSigDB) hallmark gene set collection". Cell Systems. 1 (6): 417–425. doi:10.1016/j.cels.2015.12.004. PMC 4707969. PMID 26771021.
- "Molecular signature database (MSigDB) 3.0 (PDF Download Available)". ResearchGate.
- Tamayo P, Steinhardt G, Liberzon A, Mesirov JP (February 2016). "The limitations of simple gene set enrichment analysis assuming gene independence". Statistical Methods in Medical Research. 25 (1): 472–487. arXiv:1110.4128. doi:10.1177/0962280212460441. PMC 3758419. PMID 23070592.
- Frost HR, Li Z, Moore JH (March 2015). "Spectral gene set enrichment (SGSE)". BMC Bioinformatics. 16 (1): 70. doi:10.1186/s12859-015-0490-7. PMC 4365810. PMID 25879888.
- Yousif A, Drou N, Rowe J, Khalfan M, Gunsalus KC (June 2020). "NASQAR: a web-based platform for high-throughput sequencing data analysis and visualization". BMC Bioinformatics. 21 (1): 267. bioRxiv 10.1101/709980. doi:10.1186/s12859-020-03577-4. PMC 7322916. PMID 32600310.
- "NASQAR: Nucleic Acid SeQuence Analysis Resource".
- Yu G, Wang LG, Han Y, He QY (May 2012). "clusterProfiler: an R package for comparing biological themes among gene clusters". Omics. 16 (5): 284–287. doi:10.1089/omi.2011.0118. PMC 3339379. PMID 22455463.
- "Bioconductor Org.Db Packages".
- "PlantRegMap: Plant Regulation Data and Analysis Platform @ CBI, PKU". plantregmap.cbi.pku.edu.cn.
- "GSEA | Desktop Tutorial". software.broadinstitute.org.
- "WebGestalt (WEB-based GEne SeT AnaLysis Toolkit)". www.webgestalt.org.
- "Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma'ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013; 128(14)".
- "Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma'ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research. 2016; gkw377".
- "Xie Z, Bailey A, Kuleshov MV, Clarke DJB., Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, Jeon M, & Ma'ayan A. Gene set knowledge discovery with Enrichr. Current Protocols. 1 e90 2021".
- "Ma'ayan Laboratory - Computational Systems Biology - Icahn School of Medicine at Mount Sinai". labs.icahn.mssm.edu. 19 September 2023.
- Subhash S, Kanduri C (September 2016). "GeneSCF: a real-time based functional enrichment tool with support for multiple organisms". BMC Bioinformatics. 17 (1): 365. doi:10.1186/s12859-016-1250-z. PMC 5020511. PMID 27618934.
- Wadi L, Meyer M, Weiser J, Stein LD, Reimand J (August 2016). "Impact of outdated gene annotations on pathway enrichment analysis". Nature Methods. 13 (9): 705–706. doi:10.1038/nmeth.3963. PMC 7802636. PMID 27575621. S2CID 19548133.
- "GeneSCF::Gene Set Clustering based on Functional annotation". genescf.kandurilab.org.
- "Gene Set Clustering based on Functional annotation (GeneSCF)". www.biostars.org.
- Huang DA, Sherman BT, Lempicki RA (2009). "Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources". Nature Protocols. 4 (1): 44–57. doi:10.1038/nprot.2008.211. PMID 19131956. S2CID 10418677.
- DAVID release and version information, DAVID Bioinformatics Resources 6.8
- Huang DA, Sherman BT, Lempicki RA (1 December 2008). "Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources". Nature Protocols. 4 (1): 44–57. doi:10.1038/nprot.2008.211. PMID 19131956. S2CID 10418677.
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. (April 2019). "Metascape provides a biologist-oriented resource for the analysis of systems-level datasets". Nature Communications. 10 (1): 1523. Bibcode:2019NatCo..10.1523Z. doi:10.1038/s41467-019-09234-6. PMC 6447622. PMID 30944313.
- "Metascape". metascape.org. Retrieved 20 December 2019.
- Gene Ontology Consortium. "AmiGO 2: Welcome". amigo.geneontology.org.
- Gene Ontology, Consortium (January 2015). "Gene Ontology Consortium: going forward". Nucleic Acids Research. 43 (Database issue): D1049–D1056. doi:10.1093/nar/gku1179. PMC 4383973. PMID 25428369.
- "GREAT Input: Genomic Regions Enrichment of Annotations Tool, Bejerano Lab, Stanford University". bejerano.stanford.edu.
- "GREAT improves functional interpretation of cis-regulatory regions" (PDF).
- "FunRich :: Download". funrich.org.
- Pathan M, Keerthikumar S, Ang CS, Gangoda L, Quek CY, Williamson NA, et al. (August 2015). "FunRich: An open access standalone functional enrichment and interaction network analysis tool". Proteomics. 15 (15): 2597–2601. doi:10.1002/pmic.201400515. PMID 25921073. S2CID 28583044.
- Berriz GF, Beaver JE, Cenik C, Tasan M, Roth FP (November 2009). "Next generation software for functional trend analysis". Bioinformatics. 25 (22): 3043–3044. doi:10.1093/bioinformatics/btp498. PMC 2800365. PMID 19717575.
- "List enrichment widgets statistics — InterMine documentation".
- Chen J, Bardes EE, Aronow BJ, Jegga AG (July 2009). "ToppGene Suite for gene list enrichment analysis and candidate gene prioritization". Nucleic Acids Research. 37 (Web Server issue): W305–W311. doi:10.1093/nar/gkp427. PMC 2703978. PMID 19465376.
- Yaari G, Bolen CR, Thakar J, Kleinstein SH (October 2013). "Quantitative set analysis for gene expression: a method to quantify gene set differential expression including gene-gene correlations". Nucleic Acids Research. 41 (18): e170. doi:10.1093/nar/gkt660. PMC 3794608. PMID 23921631.
- Turner JA, Bolen CR, Blankenship DM (August 2015). "Quantitative gene set analysis generalized for repeated measures, confounder adjustment, and continuous covariates". BMC Bioinformatics. 16: 272. doi:10.1186/s12859-015-0707-9. PMC 4551517. PMID 26316107.
- Avey, Stefan; et al. (August 2017). "Multicohort analysis reveals baseline transcriptional predictors of influenza vaccination responses". Science Immunology. 2 (14): eaal4656. doi:10.1126/sciimmunol.aal4656. PMC 5800877. PMID 28842433.
- Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M (September 2005). "Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research". Bioinformatics. 21 (18): 3674–3676. doi:10.1093/bioinformatics/bti610. PMID 16081474.
- "Figure 3: Heatmaps of gene set enrichment analysis (GSEA) of DEGs based on RNAseq data in response to abiotic stresses". www.nature.com. Retrieved 2018-09-05.
- Holden M, Deng S, Wojnowski L, Kulle B (December 2008). "GSEA-SNP: applying gene set enrichment analysis to SNP data from genome-wide association studies". Bioinformatics. 24 (23): 2784–2785. doi:10.1093/bioinformatics/btn516. PMID 18854360.
- Manuck TA, Watkins S, Esplin MS, Parry S, Zhang H, Huang H, Biggio JR, Bukowski R, Saade G, Andrews W, Baldwin D (2016). "242: Gene set enrichment investigation of maternal exome variation in spontaneous preterm birth (SPTB)". American Journal of Obstetrics and Gynecology. 214 (1): S142–S143. doi:10.1016/j.ajog.2015.10.280.
- Maruschke M, Hakenberg OW, Koczan D, Zimmermann W, Stief CG, Buchner A (January 2014). "Expression profiling of metastatic renal cell carcinoma using gene set enrichment analysis". International Journal of Urology. 21 (1): 46–51. doi:10.1111/iju.12183. PMID 23634695. S2CID 33377555.
- Hass J, Walton E, Wright C, Beyer A, Scholz M, Turner J, et al. (June 2015). "Associations between DNA methylation and schizophrenia-related intermediate phenotypes - a gene set enrichment analysis". Progress in Neuro-Psychopharmacology & Biological Psychiatry. 59: 31–39. doi:10.1016/j.pnpbp.2015.01.006. PMC 4346504. PMID 25598502.
- Elovainio M, Taipale T, Seppälä I, Mononen N, Raitoharju E, Jokela M, et al. (December 2015). "Activated immune-inflammatory pathways are associated with long-standing depressive symptoms: Evidence from gene-set enrichment analyses in the Young Finns Study". Journal of Psychiatric Research. 71: 120–125. doi:10.1016/j.jpsychires.2015.09.017. PMID 26473696.
Further reading
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. (October 2005). "Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles". Proceedings of the National Academy of Sciences of the United States of America. 102 (43): 15545–15550. doi:10.1073/pnas.0506580102. PMC 1239896. PMID 16199517.
- Reimand J, Isserlin R, Voisin V, Kucera M, Tannus-Lopes C, Rostamianfar A, et al. (February 2019). "Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap". Nature Protocols. 14 (2): 482–517. doi:10.1038/s41596-018-0103-9. PMC 6607905. PMID 30664679.
- Chicco D, Agapito G (August 2022). "Nine quick tips for pathway enrichment analysis". PLOS Computational Biology. 18 (8): e1010348. doi:10.1371/journal.pcbi.1010348. PMC 9371296. PMID 35951505. S2CID 251494694.