Gunther disease
Gunther disease is a congenital form of erythropoietic porphyria. The word porphyria originated from the Greek word porphura. Porphura actually means "purple pigment", which, in suggestion, the color that the body fluid changes when a person has Gunther's disease.[3] It is a rare, autosomal recessive[4] metabolic disorder affecting heme, caused by deficiency of the enzyme uroporphyrinogen cosynthetase.[5] It is extremely rare, with a prevalence estimated at 1 in 1,000,000 or less.[6] There have been times that prior to birth of a fetus, Gunther's disease has been shown to lead to anemia. In milder cases patients have not presented any symptoms until they have reached adulthood. In Gunther's disease, porphyrins are accumulated in the teeth and bones and an increased amount are seen in the plasma, bone marrow, feces, red blood cells, and urine.[7][8]
| Gunther disease | |
|---|---|
| Other names | Congenital erythropoietic porphyria (CEP), Uroporphyrinogen III synthase deficiency and UROS deficiency[1][2] |
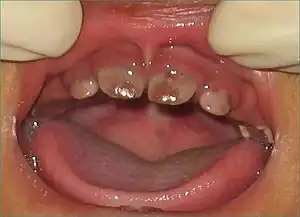 | |
| Brown discoloration of the teeth caused by porphyrin accumulation which will fluoresce under Wood's lamp (Erythrodontia). | |
| Specialty | Hematology, dermatology |
Symptoms and signs
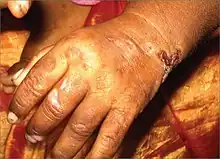
Though expressivity is varied depending on the mutation responsible for decrease in enzyme function, severe cutaneous sensitivity is present in most cases of this Porphyria. An estimated 30–40% of cases are due to the C73R mutation, which decreases stability of the enzyme and results in <1% of its activity.[9] Exposure to long-wave ultraviolet light causes the affected skin to thicken and produce vesicles that are prone to rupture and infection; these secondary infections, along with bone resorption, can lead to disfigurement of the sun-exposed face and extremities.[10] Enzyme dysfunction prevents the normal production of heme and hemolytic anemia is another common symptom, though a lack of hemolysis in this disease is possible. Porphyrins additionally accumulate in the bone and teeth, resulting in erythrodontia.[10][11] When unexpected attacks occur, abdominal pain, as well as vomiting and constipation commonly follow the attacks. Exposure to the sunlight can cause discomfort and result in blistering, consciousness of heat, and swelling and redness of the skin.[12]
Causes

Gunther disease is caused by mutations in the gene that encodes the enzyme uroporphyrinogen III synthase (UROS), located at human chromosome 10q25.2-q26.3.[4][14] The disorder is inherited in an autosomal recessive manner.[4] This means the defective gene is responsible for the disorder and is located on an autosome, and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder. When there is a homozygous mutation it causes a uroporphyrinogen III synthase and uroporphyrinogen cosynthase defect. When the enzyme uroporphyrinogen III synthase is reacting normally it results in the making of isomer III porphyrinogen, which is what is used to form heme. When isomer III porphyrinogen is not produced because of a poor production of uroporphyrinogen III synthase then isomer I porphyrinogen is made which will oxidize and give a reddish tint skin.[15][16]
Diagnosis
When diagnosing Congenital Erythroipoetic Porphyria (Gunther Disease) one must exclude other forms of porphyria. These include Hepatoerythropoietic Porphyria and rare homozygous variants of Variegate Porphyria, Hereditary Coproporphyria, and STING-associated vasculopathy with onset in Infancy (SAVI). Mild variants may be present similarly to Porphyria Cutanea Tarda.
There are four steps in establishing the diagnosis of any porphyria. First, a thorough history (particularly family history) and physical exam are performed with special attention paid to sun-exposed skin. Biochemical measurements of porphyrins and precursors in the urine, feces, and blood are necessary. Specialized labs are helpful in measuring activity of specific enzymes of the heme synthesis pathway and/or DNA and mutational analyses. In CEP, activity of uroporphyrinogen III synthase (the fourth enzyme in the heme biosynthetic pathway) will be markedly decreased.
In congenital erythropoietic porphyria, urine aminolavulanic acid and porphobilinogen are typically normal. However, uroporphyrin and coproporphyrin tend to be markedly elevated and moderately elevated, respectively, in the urine more than in the feces. Additionally, fecal protoporphyrin is typically mildly elevated.
In the red blood cells, uroporphyrin, coproporphyrin and protoporphyrin are all elevated, distinguishing this form of porphyria from the others. Finally, plasma analysis will demonstrate elevated uroporphyrin and coproporphyrin.
Other nonspecific but helpful diagnostic clues are history of cutaneous photosensitivity, blistering, erosions, crusts and ulcerations leading to extensive scarring and deformation of the hands, loss of eyebrows, eyelashes with severe mutilation of cartilaginous structures like the nose, erythrodontia, and variable degree of hematologic involvement ranging from mild hemolytic anemia to intrauterine hydrops fetalis. Other early clues are red or violet staining of diapers.
Treatment
There are a multiple ways to treat Gunther's diseases, but one of the most crucial things that a person with this disease can do is limit themselves from sun exposure or eliminate sun exposure altogether. There are some sunscreens that have undesirable effects such as tropical sunscreens, but other sunscreens containing zinc oxide and titanium dioxide are shown to provide protection due to those light-reflective agents. To block the ultraviolet and visible light wavelengths and get the protection that patients with Gunther's disease require, physical barriers are needed. It is also advised that patients wear protective clothing to block the sun from their skin. Plastic films can be attached to car windows and homes to filter out some of the wavelengths that could cause harm to someone's skin with this disease. Incandescent bulbs replace the normal fluorescent lamps. These bulbs release less light, which prevents the "porphyrin-exciting" wavelengths that fluorescent lights emit.[17]
Other less beneficial treatments have been used to help treat Gunther's disease. These include oral beta-carotene and other treatments such as activated charcoal and cholestyramine, which are used to interrupt and stop the porphyrins from being reabsorbed in the body. The reason that these oral treatments are unreasonable is because they require an extremely large dose of medicine and therefore are not beneficial.[17]
Erythrocyte transfusions have been shown to be a successful measure in decreasing the appearance of the disease by trying to lower the erythropoiesis and circulating porphyrin levels. Unfortunately, having chronic erythrocyte transfusions, it can be extremely harmful to the body and can cause severe complications.[17]
To help with dry eye symptoms and visual function, topical lubrication can be used. A more invasive way to help treat Gunther's disease would be to have surgery. There have been numerous studies that have stated that bone marrow transplantation is successful.[18] This is a recently new development for Gunther's disease so the long-term effects are still unresourced. If a patient has a life-threatening infectious complication then bone marrow transplantation is no longer relevant for them. There are also reports that stem cell transplantation is successful in a limited number of participants[17]
Eponym
The disorder is named after the German physician who discovered it, Hans Günther (1884-1956).[19]
See also
References
- Online Mendelian Inheritance in Man (OMIM): 263700
- James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. p. 526. ISBN 978-0-7216-2921-6.
- King, Michael. "Introduction to Congenital Erythropoietic Porphyria". Medical Biochemistry Page. Retrieved 30 November 2012.
- Deybach JC, De Verneuil H, Boulechfar S, Grandchamp B, Nordmann Y (1990). "Point mutations in the uroporphyrinogen III synthase gene in congenital erythropoietic porphyria (Gunther's disease)". Blood. 75 (9): 1763–5. doi:10.1182/blood.V75.9.1763.1763. ISSN 0006-4971. PMID 2331520.
- Robert-Richard E, Moreau-Gaudry F, Lalanne M, et al. (January 2008). "Effective gene therapy of mice with congenital erythropoietic porphyria is facilitated by a survival advantage of corrected erythroid cells". Am. J. Hum. Genet. 82 (1): 113–24. doi:10.1016/j.ajhg.2007.09.007. PMC 2253957. PMID 18179890.
- Thadani, H.; Deacon, A.; Peters, T. (2000). "Diagnosis and management of porphyria". BMJ. 320 (7250): 1647–1651. doi:10.1136/bmj.320.7250.1647. PMC 1127427. PMID 10856069.
- Burzio, Chiara. "Gunther's Disease". Retrieved November 28, 2012.
- Gorchein, Abel; Rong Guo; Chang Kee Lim; Ana Raimundo; Humphrey W.H. Pullon; Alastair J. Bellinham (18 January 1999). "Porphyrins in urine, plasma, erythrocytes, bile and faeces in a case of congenital erythropoietic porphyria (Gunther's disease) treated with blood transfusion and iron chelation: lack of benefit from oral charcoal". Biomedical Chromatography. 12 (6): 350–356. doi:10.1002/(SICI)1099-0801(199811/12)12:6<350::AID-BMC761>3.0.CO;2-B. PMID 9861496.
- Fortian, A; González, E; Castaño, D; Falcon-Perez, JM; Millet, O (Apr 15, 2011). "Intracellular rescue of the uroporphyrinogen III synthase activity in enzymes carrying the hotspot mutation C73R". The Journal of Biological Chemistry. 286 (15): 13127–33. doi:10.1074/jbc.m110.205849. PMC 3075659. PMID 21343304.
- Balwani, M; Desnick, RJ (2012). "The porphyrias: Advances in diagnosis and treatment". Hematology. 2012: 19–27. doi:10.1182/asheducation.v2012.1.19.3795678. PMID 23233556.
- De, AK; Das, K; Sil, A; Joardar, S (Sep 2013). "A Case of Congenital Erythropoietic Porphyria without Hemolysis". Indian Journal of Dermatology. 58 (5): 407. doi:10.4103/0019-5154.117336. PMC 3778801. PMID 24082206.
- Saval, Herrera; Moruno Tirado (24 December 2001). "Congenital erythropoietic porphyria affecting two brothers". British Journal of Dermatology. 141 (3): 547–550. doi:10.1046/j.1365-2133.1999.03057.x. PMID 10583066.
- Katugampola, RP; Anstey, AV; Finlay, AY; Whatley, S; Woolf, J; Mason, N; Deybach, JC; Puy, H; Ged, C; de Verneuil, H; Hanneken, S; Minder, E; Schneider-Yin, X; Badminton, MN (Oct 2012). "A management algorithm for congenital erythropoietic porphyria derived from a study of 29 cases". The British Journal of Dermatology. 167 (4): 888–900. doi:10.1111/j.1365-2133.2012.11154.x. PMID 22804244.
- Online Mendelian Inheritance in Man (OMIM): 606938
- "Congenital erythropoietic porphyria". New Zealand Dermatological Society. Retrieved 28 November 2012.
- Pannier, E; G. Viot; M.C. Aubry; G. Grange; J. Tantau; C. Fallet-Bianco; F. Muller; D. Cabrol (9 December 2002). "Congenital erythropoietic porphyria (Günther's disease): two cases with very early prenatal manifestation and cystic hygroma". Prenatal Diagnosis. 23 (1): 25–30. doi:10.1002/pd.491. PMID 12533808. S2CID 25995061.
- Hebel, Jeanette (2018-05-24). "Congenital Erythropoietic Porphyria Treatment & Management". Medscape. Retrieved 30 November 2012.
- Taibjee, S.M; O.E. Steveson; A. Adullah; C.Y. Tan; P. Darbyshire; C. Moss; H. Goodyear; A. Heargerty; S. Wheatley; M.N. Badminton (18 January 2007). "Allogeneic bone marrow transplantation in a 7-year-old girl with congenital erythropoietic porphyria: a treatment dilemma". British Journal of Dermatology. 156 (3): 567–571. doi:10.1111/j.1365-2133.2006.07699.x. PMID 17300251. S2CID 39733402.
- Madan P, Schaaf CP, Vardhan P, Bhayana S, Chandra P, Anderson KE (2007). "Hans Gunther and his disease". Photodermatol Photoimmunol Photomed. 23 (6): 261–3. doi:10.1111/j.1600-0781.2007.00323.x. PMID 17986065. S2CID 44372076.
External links
- Porphyria, congenital erythropoietic at NIH's Office of Rare Diseases