HEXA
Hexosaminidase A (alpha polypeptide), also known as HEXA, is an enzyme that in humans is encoded by the HEXA gene, located on the 15th chromosome.[5][6]
| HEXA | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Identifiers | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Aliases | HEXA, TSD, hexosaminidase subunit alpha | ||||||||||||||||||||||||||||||||||||||||||||||||||
| External IDs | OMIM: 606869 MGI: 96073 HomoloGene: 20146 GeneCards: HEXA | ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||




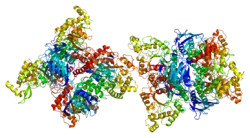
Hexosaminidase A and the cofactor GM2 activator protein catalyze the degradation of the GM2 gangliosides and other molecules containing terminal N-acetyl hexosamines.[7] Hexosaminidase A is a heterodimer composed of an alpha subunit (this protein) and a beta subunit. The alpha subunit polypeptide is encoded by the HEXA gene while the beta subunit is encoded by the HEXB gene. Gene mutations in the gene encoding the beta subunit (HEXB) often result in Sandhoff disease; whereas, mutations in the gene encoding the alpha subunit (HEXA, this gene) decrease the hydrolysis of GM2 gangliosides, which is the main cause of Tay–Sachs disease.[8]
Function
Even though the alpha and beta subunits of hexosaminidase A can both cleave GalNAc residues, only the alpha subunit is able to hydrolyze GM2 gangliosides. The alpha subunit contains a key residue, Arg-424, which is essential for binding the N-acetyl-neuramanic residue of GM2 gangliosides. The alpha subunit can hydrolyze GM2 gangliosides because it contains a loop structure consisting of the amino acids: Gly-280, Ser-281, Glu-282, and Pro-283. The loop is absent in the beta subunit, but it serves as an ideal structure for the binding of the GM2 activator protein (GM2AP) in the alpha subunit. A combination of Arg-424 and the amino acids that cause the formation of the loop allow the alpha subunit to hydrolyze GM2 gangliosides into GM3 gangliosides by removing the N-acetylgalactosamine (GalNAc) residue from GM2 gangliosides.[9]
Gene mutations resulting in Tay–Sachs disease
There are numerous mutations that lead to hexosaminidase A deficiency including gene deletions, nonsense mutations, and missense mutations. Tay–Sachs disease occurs when hexosaminidase A loses its ability to function. People with Tay–Sachs disease are unable to remove the GalNAc residue from the GM2 ganglioside, and as a result, they end up storing 100 to 1000 times more GM2 gangliosides in the brain than the normal person. Over 100 different mutations have been discovered just in infantile cases of Tay–Sachs disease alone.[10]
The most common mutation, which occurs in over 80 percent of Tay–Sachs patients, results from a four base pair addition (TATC) in exon 11 of the Hex A gene. This insertion leads to an early stop codon, which causes the Hex A deficiency.[11]
Children born with Tay–Sachs usually die between two and six years of age from aspiration and pneumonia. Tay–Sachs causes cerebral degeneration and blindness. Patients also experience flaccid extremities and seizures. There is no cure for Tay–Sachs disease.[10]
Gene Therapies for Tay-Sachs
The HEXA gene is a protein encoding gene that codes for the lysosomal enzyme beta-hexosaminidase. This enzyme, combined with the GM2 activator protein, is responsible for the breakdown of ganglioside GM2 within the lysosome. Defects in the HEXA gene, however, prevent this degradation, leading to a buildup of toxins in brain and spinal cord cells. This fatal genetic disorder is called Tay-Sachs disease. Because the Tay-Sachs gene defect mainly affects neural cells, a patient with the HEXA mutation will experience a quick deterioration of motor and mental function before dying around the age of three or four. [8]
A “knockout” model, which is a mouse that has been genetically modified to observe the effects of inactivation of or damage to certain genes, found that the mice that were administered the HEXA gene experienced many of the same symptoms of Tay-Sachs, with one exception: GM2 buildup was distributed differently in the brains of the mice than in those of a typical human Tay-Sachs patient. [9] This model has allowed scientists to research gene therapies for HEXA defects. One study, done on mice, successfully reestablished beta-hexoaminidase levels and removed the toxic cell buildup by using a non-replicated Herpes simplex vector to code for the missing gene. [10]
References
- GRCh38: Ensembl release 89: ENSG00000213614 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000025232 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Korneluk RG, Mahuran DJ, Neote K, Klavins MH, O'Dowd BF, Tropak M, Willard HF, Anderson MJ, Lowden JA, Gravel RA (June 1986). "Isolation of cDNA clones coding for the alpha-subunit of human beta-hexosaminidase. Extensive homology between the alpha- and beta-subunits and studies on Tay-Sachs disease". The Journal of Biological Chemistry. 261 (18): 8407–13. doi:10.1016/S0021-9258(19)83927-3. PMID 3013851.
- Proia RL, Soravia E (April 1987). "Organization of the gene encoding the human beta-hexosaminidase alpha-chain". The Journal of Biological Chemistry. 262 (12): 5677–81. doi:10.1016/S0021-9258(18)45628-1. PMID 2952641.
- Knapp S, Vocadlo D, Gao Z, Kirk B, Lou J, Withers SG (1996). "NAG-thiazoline, an N-acetylbeta-hexosaminidase inhibitor that implicates acetamido participation". J. Am. Chem. Soc. 118 (28): 6804–6805. doi:10.1021/ja960826u.
- Mark BL, Mahuran DJ, Cherney MM, Zhao D, Knapp S, James MN (April 2003). "Crystal structure of human beta-hexosaminidase B: understanding the molecular basis of Sandhoff and Tay-Sachs disease". Journal of Molecular Biology. 327 (5): 1093–109. doi:10.1016/S0022-2836(03)00216-X. PMC 2910754. PMID 12662933.
- Lemieux MJ, Mark BL, Cherney MM, Withers SG, Mahuran DJ, James MN (June 2006). "Crystallographic structure of human beta-hexosaminidase A: interpretation of Tay-Sachs mutations and loss of GM2 ganglioside hydrolysis". Journal of Molecular Biology. 359 (4): 913–29. doi:10.1016/j.jmb.2006.04.004. PMC 2910082. PMID 16698036.
- Ozand PT, Nyhan WL, Barshop BA (2005). "Part Thirteen Lipid Storage Disorders: Tay-Sachs disease/hexosaminidase A deficiency". Atlas of metabolic diseases. London: Hodder Arnold. pp. 539–546. ISBN 0-340-80970-1.
- Boles DJ, Proia RL (March 1995). "The molecular basis of HEXA mRNA deficiency caused by the most common Tay-Sachs disease mutation". American Journal of Human Genetics. 56 (3): 716–24. PMC 1801160. PMID 7887427.
Further reading
- Taniike, Masako; Yamanaka, Shoji; Proia, Richard L.; Langaman, Clarita; Bone-Turrentine, Teresa; Suzuki, Kinuko (1995). "Neuropathology of mice with targeted disruption of Hexa gene, a model of Tay-Sachs disease". Acta Neuropathologica. 89 (4): 296–304. doi:10.1007/s004010050250. PMID 7610760.
- Martino S, Marconi P, Tancini B, Dolcetta D, De Angelis MG, Montanucci P, Bregola G, Sandhoff K, Bordignon C, Emiliani C, Manservigi R, Orlacchio A (August 2005). "A direct gene transfer strategy via brain internal capsule reverses the biochemical defect in Tay-Sachs disease". Human Molecular Genetics. 14 (15): 2113–23. doi:10.1093/hmg/ddi216. PMID 15961412.
- Mahuran DJ (February 1991). "The biochemistry of HEXA and HEXB gene mutations causing GM2 gangliosidosis". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1096 (2): 87–94. doi:10.1016/0925-4439(91)90044-A. PMID 1825792.
- Myerowitz R (1997). "Tay-Sachs disease-causing mutations and neutral polymorphisms in the Hex A gene". Human Mutation. 9 (3): 195–208. doi:10.1002/(SICI)1098-1004(1997)9:3<195::AID-HUMU1>3.0.CO;2-7. PMID 9090523. S2CID 22587938.
- Mahuran DJ (October 1999). "Biochemical consequences of mutations causing the GM2 gangliosidoses". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1455 (2–3): 105–38. doi:10.1016/S0925-4439(99)00074-5. PMID 10571007.
- Gilbert F, Kucherlapati R, Creagan RP, Murnane MJ, Darlington GJ, Ruddle FH (January 1975). "Tay-Sachs' and Sandhoff's diseases: the assignment of genes for hexosaminidase A and B to individual human chromosomes". Proceedings of the National Academy of Sciences of the United States of America. 72 (1): 263–7. doi:10.1073/pnas.72.1.263. PMC 432284. PMID 1054503.
- Trop I, Kaplan F, Brown C, Mahuran D, Hechtman P (1993). "A glycine250--> aspartate substitution in the alpha-subunit of hexosaminidase A causes juvenile-onset Tay-Sachs disease in a Lebanese-Canadian family". Human Mutation. 1 (1): 35–9. doi:10.1002/humu.1380010106. PMID 1301189. S2CID 40974834.
- Akalin N, Shi HP, Vavougios G, Hechtman P, Lo W, Scriver CR, Mahuran D, Kaplan F (1993). "Novel Tay-Sachs disease mutations from China". Human Mutation. 1 (1): 40–6. doi:10.1002/humu.1380010107. PMID 1301190. S2CID 28734677.
- Akerman BR, Zielenski J, Triggs-Raine BL, Prence EM, Natowicz MR, Lim-Steele JS, Kaback MM, Mules EH, Thomas GH, Clarke JT (1993). "A mutation common in non-Jewish Tay-Sachs disease: frequency and RNA studies". Human Mutation. 1 (4): 303–9. doi:10.1002/humu.1380010407. PMID 1301938. S2CID 25926877.
- Fernandes M, Kaplan F, Natowicz M, Prence E, Kolodny E, Kaback M, Hechtman P (December 1992). "A new Tay-Sachs disease B1 allele in exon 7 in two compound heterozygotes each with a second novel mutation". Human Molecular Genetics. 1 (9): 759–61. doi:10.1093/hmg/1.9.759. PMID 1302612.
- McDowell GA, Mules EH, Fabacher P, Shapira E, Blitzer MG (November 1992). "The presence of two different infantile Tay-Sachs disease mutations in a Cajun population". American Journal of Human Genetics. 51 (5): 1071–7. PMC 1682822. PMID 1307230.
- Whitley CB, Anderson RA, McIvor RS (April 1992). "Heterozygosity for the "DN allele" (G533-greater than A) of the beta-hexosaminidase alpha subunit gene identified by direct DNA sequencing in a family with the B1 variant of GM2-gangliosidosis". Neuropediatrics. 23 (2): 96–101. doi:10.1055/s-2008-1071320. PMID 1318511.
- Triggs-Raine BL, Mules EH, Kaback MM, Lim-Steele JS, Dowling CE, Akerman BR, Natowicz MR, Grebner EE, Navon R, Welch JP (October 1992). "A pseudodeficiency allele common in non-Jewish Tay-Sachs carriers: implications for carrier screening". American Journal of Human Genetics. 51 (4): 793–801. PMC 1682803. PMID 1384323.
- Hechtman P, Boulay B, De Braekeleer M, Andermann E, Melançon S, Larochelle J, Prevost C, Kaplan F (December 1992). "The intron 7 donor splice site transition: a second Tay-Sachs disease mutation in French Canada". Human Genetics. 90 (4): 402–6. doi:10.1007/bf00220467. PMID 1483696. S2CID 3190945.
- Mules EH, Hayflick S, Miller CS, Reynolds LW, Thomas GH (April 1992). "Six novel deleterious and three neutral mutations in the gene encoding the alpha-subunit of hexosaminidase A in non-Jewish individuals". American Journal of Human Genetics. 50 (4): 834–41. PMC 1682641. PMID 1532289.
- Weitz G, Proia RL (May 1992). "Analysis of the glycosylation and phosphorylation of the alpha-subunit of the lysosomal enzyme, beta-hexosaminidase A, by site-directed mutagenesis". The Journal of Biological Chemistry. 267 (14): 10039–44. doi:10.1016/S0021-9258(19)50196-X. PMID 1533633.
- Navon R, Proia RL (February 1991). "Tay-Sachs disease in Moroccan Jews: deletion of a phenylalanine in the alpha-subunit of beta-hexosaminidase". American Journal of Human Genetics. 48 (2): 412–9. PMC 1683003. PMID 1825014.
- Mules EH, Dowling CE, Petersen MB, Kazazian HH, Thomas GH (June 1991). "A novel mutation in the invariant AG of the acceptor splice site of intron 4 of the beta-hexosaminidase alpha-subunit gene in two unrelated American black GM2-gangliosidosis (Tay-Sachs disease) patients". American Journal of Human Genetics. 48 (6): 1181–5. PMC 1683116. PMID 1827945.
- Nakai H, Byers MG, Nowak NJ, Shows TB (1991). "Assignment of beta-hexosaminidase A alpha-subunit to human chromosomal region 15q23----q24". Cytogenetics and Cell Genetics. 56 (3–4): 164. doi:10.1159/000133077. PMID 1829032.
- Nishimoto J, Tanaka A, Nanba E, Suzuki K (August 1991). "Expression of the beta-hexosaminidase alpha subunit gene with the four-base insertion of infantile Jewish Tay-Sachs disease". The Journal of Biological Chemistry. 266 (22): 14306–9. doi:10.1016/S0021-9258(18)98684-9. PMID 1830584.
- dos Santos MR, Tanaka A, sá Miranda MC, Ribeiro MG, Maia M, Suzuki K (October 1991). "GM2-gangliosidosis B1 variant: analysis of beta-hexosaminidase alpha gene mutations in 11 patients from a defined region in Portugal". American Journal of Human Genetics. 49 (4): 886–90. PMC 1683169. PMID 1832817.
External links
- Hexosaminidase A at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- EC 3.2.1.52
- National Tay-Sach’s Disease Site
PDB gallery | |
|---|---|
|

