Hot start PCR
Hot start PCR is a modified form of conventional polymerase chain reaction (PCR) that reduces the presence of undesired products and primer dimers due to non-specific DNA amplification at room (or colder) temperatures.[1][2] Many variations and modifications of the PCR procedure have been developed in order to achieve higher yields; hot start PCR is one of them.[3] Hot start PCR follows the same principles as the conventional PCR - in that it uses DNA polymerase to synthesise DNA from a single stranded template.[4] However, it utilizes additional heating and separation methods, such as inactivating or inhibiting the binding of Taq polymerase and late addition of Taq polymerase, to increase product yield as well as provide a higher specificity and sensitivity.[5] Non-specific binding and priming or formation of primer dimers are minimized by completing the reaction mix after denaturation.[6] Some ways to complete reaction mixes at high temperatures involve modifications that block DNA polymerase activity in low temperatures,[1][7] use of modified deoxyribonucleotide triphosphates (dNTPs),[8] and the physical addition of one of the essential reagents after denaturation.[9]
Through these additional methods, hot start PCR is able to decrease the amount of non-specific amplifications which naturally occur during lower temperatures – which remains a problem for conventional PCR. These modifications work overall to ensure that specific enzymes in solution will remain inactive or are inhibited until the optimal annealing temperature is reached.[10] Inhibiting formation of non-specific PCR products, especially in early cycles, results in a substantial increase in sensitivity of amplification by PCR. This is of utmost importance in diagnostic applications of PCR or RT-PCR.
Background
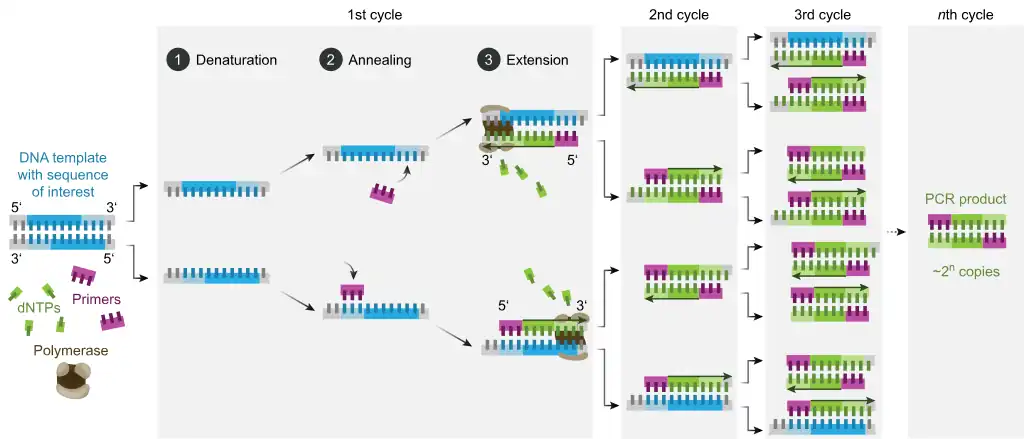
Polymerase chain reaction (PCR) is a molecular biology technique used to amplify specific DNA segments by several orders of magnitude. The specific segments of DNA is amplified over three processes, denaturation, annealing and extension – where the DNA strands are separated by raising the temperature to the optimal from room temperature before primers bind and polymerase aligns nucleotides to the template strand. It uses DNA polymerase, which is slightly active at low temperatures.[1] In conventional PCR, the reaction mix is completed at room temperature, and due to DNA polymerase activity, primers may form primer dimers or anneal to DNA non-specifically. During the PCR procedure, DNA polymerase will extend any piece of DNA with bound primers, generating target products but also nonspecific products which lower the yield. In hot start PCR, some of the reagents are kept separate until the mixture is heated to the specific annealing temperature. This reduces annealing time, which in turn reduces the likelihood of non-specific DNA extension and the influence of non-specific primer binding prior to denaturation.[6][5]
In conventional PCR, lower temperatures below the optimal annealing temperature (50-65 °C) results in off target modifications such as non-specific amplifications where primers will bind non-specifically to the nucleic acid.[5] These non-specific primer complexes, which are in excess in the mixture, are the cause behind the synthesis of by-products such as primer dimer and mis-priming.[10] Mis-priming greatly impedes and reduces the efficiency of PCR amplification through actively competing with the target sequences for amplification. Similarly, primer dimers form complexes which decreases the amount of copy number amplifications obtained.[10] This can be controlled by implementing hot start PCR which allows primer extensions to be blocked until the optimal temperatures are met.[2]
In hot start PCR, important reagents (such as DNA polymerase and magnesium cofactors) are prevented from reacting in the PCR mixture until the optimal temperatures are met through physical separation or chemical modifications.[5][2] Hot start PCR can also occur when the Taq polymerase is inhibited/inactivated or its addition is delayed until optimal annealing temperatures, through deoxyribonucleotide triphosphate modifications or by modifying the primers through caging and secondary structure manipulation.
Hot start PCR is often a better approach opposed to traditional PCR in circumstances where there is a low concentration of DNA in the reaction mix, the DNA template is highly complex, or if there are several pairs of oligonucleotide primers in the PCR.[3]
Methods
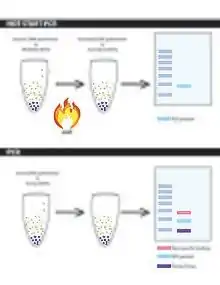
Hot start PCR is a method which prevents DNA polymerase extension at lower temperature to prevent non-specific binding to minimise yield loss. Hot start PCR reduces the amount of non-specific binding through limiting reagents until the heating steps of PCR – limit the reaction early by limiting Taq DNA polymerase in a reaction. Non-specific binding often leads to primer dimers and mis-primed/false primed targets.[11] These can be rectified through modified methods such as:
Inactivation/inhibition of Taq DNA polymerase
Enzyme linked antibodies/Taq DNA polymerase complexed with Anti Taq DNA polymerase antibodies:
The enzyme linked antibodies inactivate the Taq DNA polymerase. The antibodies link and bind to the polymerase, preventing early DNA amplification which could occur at lower temperatures. Once the optimal annealing temperature is met, the antibodies will begin to degrade and dissociate, releasing the Taq DNA polymerase into the reaction and allowing the amplification process to start.[2][12] Platinum Taq DNA polymerase and AccuStart Taq DNA polymerase ( both developed by Ayoub Rashtchian at Life technologies and Quanta BioSciences, respectively) are examples of commercially available antibody based hot start Taq DNA polymerases. These Taq DNA polymerase are precomplexed with a mixture of monoclonal antibodies specific to Taq DNA polymerase.[13]
Wax beads:
A physical barrier is created between Taq DNA polymerase and the remainder of the PCR components by the wax beads which are temperature dependent. Once the temperature rises over 70 °C, during the denaturation step in the first cycle, the wax bead melts, allowing the Taq DNA polymerase to escape past the barrier and be released into the reaction – starting the amplification process. The wax layer then moves to the top of the reaction mixture during the amplification stage to later act as a vapour barrier.[2]
Highly specific oligonucleotides:
Oligonucleotides are short polymers of nucleic acid which easily bind. Highly specific oligonucleotides, such as aptamers, bind to Taq DNA polymerase at lower temperatures making it inactive in the mixture. Only at higher temperatures will the oligonucleotides separate from the Taq allowing it to react.[5]
These are the most effective methods for hot start PCR, the enzyme linked antibodies and highly specific oligonucleotides methods in particular are most suited during procedures which require a shorter inactivation time.[14] However, other methods are known to be implemented such as:
Late addition of Taq DNA polymerase
Preheating:
The PCR machine is heated in advance whilst the components are mixed over ice and then immediately placed into the PCR machine once it reaches optimum temperature. This would eliminate the warm-up process required, reduce non-specific annealing of the primers and ensures that any miss paired primers in the mixture are separated.[15]
Freezing:
Freezing acts as a form of physical separation much like the wax beads. The reaction mixture containing primers, the template strand, water and deoxyribonucleotide triphosphate (dNTP) is frozen before Taq polymerase and the remaining PCR components are added on top of the frozen mixture. This acts to prevent non-specific binding.[15]
Later addition of Taq:
The components of PCR in the reaction mix are prepared and heated without the addition of Taq. Taq is only later introduced into the mixture once the optimal temperature is reached. However, this method is the least reliable and may lead to a contamination of the components.[15]
Another method is through deoxyribonucleotide triphosphate mediated hot start PCR which modifies the nucleotide bases through a protecting group.
Deoxyribonucleotide triphosphate (dNTP) modifications
Hot start dNTP can be chemically modified to include a heat sensitive protecting group at the 3 prime terminus. This modification will prevent the nucleotides from interacting with the Taq polymerase to bind to the template strand until after the optimal temperatures are reached therefore, the protecting group will be removed during the heat activation step. The hot start dNTP, dA, dT, dC and dG replace the natural nucleotides.[16][17] Using all four of the modified nucleotides is recommended, however, previous research shows that by replacing either one or two of the natural nucleotides with the modified dNTPs would be enough to ensure that non-specific amplification does not occur.[16][17] Another chemical modification of nucleic acid is through the heat-reversible covalent modification which acts to impede the hybridisation of the primers to the template of interest. The guanosine amino group interact with glyoxal to form dG.[18]
Modified primers
Secondary structure:
Certain secondary structure may impede the functions of the primers. For example, oligonucleotides with a hairpin structure cannot act efficiently as a primer. However, after heating the reaction mix to the annealing temperature the primer will undergo a conformation change allowing the primer to form a linear structure instead, which enables the primer to attach to the target segment and begin PCR.[19][20] Actually, there is a more stable configuration to the hairpin primers termed ‘double-bubble’ primers, that form a head to tail homodimer configuration that can be utilized both for reverse- transcription and for hot- start PCR. [21]
Photochemically removable cages:
A caging group which is a protecting group that is photochemically removable, such as caged thymidine phosphoramidites, is incorporated into a oligonucleotide primer. This allows the function of the primer to be activated and deactivated through the use of UV irradiation (365 nm). Therefore, primers can be activated after the annealing temperature is reached.[22]
Controlled addition of magnesium
Magnesium is required in PCR and acts as a co-factor because Taq polymerase is magnesium dependent.[23] Increasing the concentration of magnesium and phosphate to the standard buffer reagents creates a magnesium precipitate, providing a hot start for the reaction as there is no magnesium for the DNA polymerase until during the thermal cycling stage. During thermal cycling, the magnesium will dissolve back into solution and become available for the polymerase to use allowing it to function normally.[24]
Advantages
Hot start PCR is advantageous in that it requires less handling and reduces the risk of contamination. Hot start PCR can either be chemically modified or antibody based which provide different advantages to the procedure. In chemically modified hot start PCR, the procedure can be taken under room temperature and significantly decreases the formation of primer-dimers by preventing primers from binding to one another before the PCR process has begun as well as limiting non-specific priming. Similarly, hot start PCR inhibits the binding of primers to the template sequences which have a low homology which leads to mispriming. It can also improve specificity and sensitivity, due to the stringent conditions, as well as increase the product yield of the targeted fragment.[5] In antibody based hot start PCR, the polymerase is activated after the initial denaturation step during the cycling process, therefore decreasing the time required. This also leads to a high specificity.[14]
Limitations
Along with its advantages, hot start PCR also has limitations which must be considered before implementing the method. Hot start PCR requires the addition of heat for longer periods of time as opposed to conventional PCR, therefore, the template DNA is more susceptible to being damaged. The increased heating time also means that the procedure is not compatible for certain procedures such as the one tube, single buffer reverse transcription-PCR method which requires lower temperature to undergo the reverse transcription step.[12] In chemically modified hot start PCR, the amplification process of DNA can be negatively affected firstly due to a significant increase in the reactivation time required for the polymerase to activate and secondly if the length of the target DNA template is too long.[14] In antibody based procedures, each enzyme requires a different antibody and therefore the cost to perform the procedure is higher.[15] There is also evidence that many commercial hot start enzymes actually have some level of activity prior to denaturation, and few suppliers provide any information about testing for this residual activity.[25] This means that the benefits attributed to hot start enzymes may not be realized, or at best will vary between batches or manufacturer.
References
- Sharkey DJ, Scalice ER, Christy KG, Atwood SM, Daiss JL (May 1994). "Antibodies as thermolabile switches: high temperature triggering for the polymerase chain reaction". Bio/Technology. 12 (5): 506–9. doi:10.1038/nbt0594-506. PMID 7764710. S2CID 2885453.
- Paul N, Shum J, Le T (2010). "Hot start PCR". RT-PCR Protocols. Methods in Molecular Biology. Vol. 630. Humana Press. pp. 301–18. doi:10.1007/978-1-60761-629-0_19. ISBN 9781607616283. PMID 20301005.
- Green, Michael R.; Sambrook, Joseph (May 2018). "Hot Start Polymerase Chain Reaction (PCR)". Cold Spring Harbor Protocols. 2018 (5): pdb.prot095125. doi:10.1101/pdb.prot095125. ISSN 1940-3402. PMID 29717052.
- Aryal, Sagar (2015-04-23). "Polymerase Chain Reaction (PCR)- Principle, Procedure, Types, Applications and Animation". Microbiology Info.com. Retrieved 2020-05-29.
- Birch DE (May 1996). "Simplified hot start PCR". Nature. 381 (6581): 445–6. Bibcode:1996Natur.381..445B. doi:10.1038/381445a0. PMID 8632804. S2CID 4267296.
- van Pelt-Verkuil E, van Belkum A, Hays JP, eds. (2008). "Variants and Adaptations of the Standard PCR Protocol". Principles and Technical Aspects of PCR Amplification. Springer Netherlands. pp. 231–276. doi:10.1007/978-1-4020-6241-4_12. ISBN 9781402062414.
- "How is Hot-Start Technology Beneficial For Your PCR". Thermofisher. Retrieved 2019-10-03.
- "DNTP - The School of Biomedical Sciences Wiki". teaching.ncl.ac.uk. Retrieved 2019-10-09.
- Coleman WB (2016). Diagnostic molecular pathology. [London]: Elsevier Academic Press. ISBN 9780128011577. OCLC 960448665.
- Lebedev, Alexandre V.; Paul, Natasha; Yee, Joyclyn; Timoshchuk, Victor A.; Shum, Jonathan; Miyagi, Kei; Kellum, Jack; Hogrefe, Richard I.; Zon, Gerald (November 2008). "Hot Start PCR with heat-activatable primers: a novel approach for improved PCR performance". Nucleic Acids Research. 36 (20): e131. doi:10.1093/nar/gkn575. ISSN 1362-4962. PMC 2582603. PMID 18796527.
- Kermekchiev, Milko B.; Tzekov, Anatoly; Barnes, Wayne M. (2003-11-01). "Cold‐sensitive mutants of Taq DNA polymerase provide a hot start for PCR". Nucleic Acids Research. 31 (21): 6139–6147. doi:10.1093/nar/gkg813. ISSN 0305-1048. PMC 275455. PMID 14576300.
- Schoenbrunner, Nancy J; Gupta, Amar P; Young, Karen K Y; Will, Stephen G (2017-01-01). "Covalent modification of primers improves PCR amplification specificity and yield". Biology Methods and Protocols. 2 (1): bpx011. doi:10.1093/biomethods/bpx011. ISSN 2396-8923. PMC 6994073. PMID 32161793.
- Westfall et.al. (1997), Focus 19.3, page 46.
- "How is Hot-Start Technology Beneficial For Your PCR - AU". www.thermofisher.com. Retrieved 2020-05-29.
- "What is a hot start PCR?". Genetic Education. 2019-03-03. Retrieved 2020-05-29.
- Koukhareva, Inna; Lebedev, Alexandre (2009-06-15). "3′-Protected 2′-Deoxynucleoside 5′-Triphosphates as a Tool for Heat-Triggered Activation of Polymerase Chain Reaction". Analytical Chemistry. 81 (12): 4955–4962. doi:10.1021/ac8026977. ISSN 0003-2700. PMC 2712722. PMID 19438248.
- Koukhareva, I.; Haoqiang, H.; Yee, J.; Shum, J.; Paul, N.; Hogrefe, R. I.; Lebedev, A. V. (2008-09-01). "Heat Activatable 3'-modified dNTPs: Synthesis and Application for Hot Start PCR". Nucleic Acids Symposium Series. 52 (1): 259–260. doi:10.1093/nass/nrn131. ISSN 0261-3166. PMID 18776352.
- US application 20030162199, Bonner, Alex, "Reversible chemical modification of nucleic acids and improved method for nucleic acid hybridization", published 2003-08-28, assigned to BioLink Partners Inc., since abandoned.
- Ailenberg, M and Silverman, M, 2000-11-1, Controlled hot start and improved specificity in carrying out PCR utilizing touch-up and loop incorporated primers (TULIPS),Biotechniques, 29(5):1018- 1022,doi: 10.2144/00295st03,PMID: 11084864
- Kaboev, O. K.; Luchkina, L. A.; Tret’iakov, A. N.; Bahrmand, A. R. (2000-11-01). "PCR hot start using primers with the structure of molecular beacons (hairpin-like structure)". Nucleic Acids Research. 28 (21): e94. doi:10.1093/nar/28.21.e94. ISSN 0305-1048. PMC 113163. PMID 11058144.
- Ailenberg, M, Kapus, A and Rotstein, OD, 2021-09-14, Improved SARS-CoV-2 PCR detection and genotyping with double-bubble primers, Biotechniques,71 (6):587-597, DOI: 10.2144/btn-2021-0063
- Young, Douglas D.; Edwards, Wesleigh F.; Lusic, Hrvoje; Lively, Mark O.; Deiters, Alexander (2008-01-10). "Light-triggered polymerase chain reaction". Chemical Communications (4): 462–464. doi:10.1039/B715152G. ISSN 1364-548X. PMC 3760149. PMID 18188468.
- Markoulatos, P.; Siafakas, N.; Moncany, M. (2002). "Multiplex polymerase chain reaction: A practical approach". Journal of Clinical Laboratory Analysis. 16 (1): 47–51. doi:10.1002/jcla.2058. ISSN 0887-8013. PMC 6808141. PMID 11835531.
- Barnes, Wayne M; Rowlyk, Katherine R (June 2002). "Magnesium precipitate hot start method for PCR". Molecular and Cellular Probes. 16 (3): 167–171. doi:10.1006/mcpr.2002.0407. PMID 12219733.
- Stevens AJ, et al. (Aug 2016). "Many commercial hot-start polymerases demonstrate activity prior to thermal activation". BioTechniques. 61 (6): 293–296. doi:10.2144/000114481. PMID 25967906.