T7 DNA polymerase
T7 DNA polymerase is an enzyme used during the DNA replication of the T7 bacteriophage. During this process, the DNA polymerase “reads” existing DNA strands and creates two new strands that match the existing ones. The T7 DNA polymerase requires a host factor, E. coli thioredoxin,[1] in order to carry out its function. This helps stabilize the binding of the necessary protein to the primer-template to improve processivity by more than 100-fold, which is a feature unique to this enzyme.[2] It is a member of the Family A DNA polymerases, which include E. coli DNA polymerase I and Taq DNA polymerase.
| DNA-directed DNA polymerase | |||||||
|---|---|---|---|---|---|---|---|
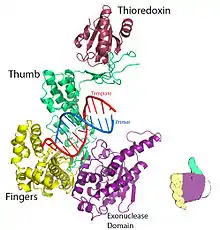 Figure 1. Crystal structure of T7 DNA replication complex. Rendered from PDB 1T7P. | |||||||
| Identifiers | |||||||
| Organism | |||||||
| Symbol | 5 | ||||||
| CAS number | 9012-90-2 | ||||||
| UniProt | P00581 | ||||||
| Other data | |||||||
| EC number | 2.7.7.7 | ||||||
| |||||||
This polymerase has various applications in site-directed mutagenesis[3] as well as a high-fidelity enzyme suitable for PCR.[4] It has also served as the precursor to Sequenase,[5] an engineered-enzyme optimized for DNA sequencing.[6]
Mechanism
Phosphoryl transfer

Figure 2. Nucleotidyl transfer by DNA polymerase.
T7 DNA polymerase catalyzes the phosphoryl transfer [7] during DNA replication of the T7 phage. As shown in Figure 2, the 3’ hydroxyl group of a primer acts as a nucleophile and attacks the phosphodiester bond of nucleoside 5’-triphosphate (dTMP-PP). This reaction adds a nucleoside monophosphate into DNA and releases a pyrophosphate (PPi). Generally, the reaction is metal-dependent and cations such as Mg2+ are often present in the enzyme active site.[7]
For T7 DNA polymerase, the fingers, palm and thumb (Figure 1) position the primer-template so that the 3’-end of the primer strand is positioned next to the nucleotide-binding site (located at the intersection of the fingers and thumb).[8] The base pair formed between the nucleotide and the template base fits nicely into a groove between the fingers and the 3’-end of the primer.[8] Two Mg2+ ions form an octahedral coordinate network with oxygen ligand and also bring the reactive primer hydroxyl and the nucleotide α-phosphate close together, thereby lowering the entropic cost of nucleophilic addition.[8] The rate-limiting step in the catalytic cycle occurs after the nucleoside triphosphate binds and before it is incorporated into the DNA (corresponding to the closure of the fingers subdomain around the DNA and nucleotide).[8]

Role of Mg2+ ions and amino acid residues in the active site
The amino acids present in the active site assist in creating a stabilizing environment for the reaction to proceed. Amino acids such as Lys522, Tyr526, His506 and Arg518 act as hydrogen bond donors. The backbone carbonyl of Ala476, Asp475 and Asp654 form coordinate bonds with the Mg2+ ions.
Asp475 and Asp654 form a bridge with the Mg2+ cations to orient them properly. The Mg2+ ion on the right (Figure 3) interacts with negatively charged oxygens of the alpha(α), beta(β) and gamma(γ) phosphates to align the scissile bond for the primer to attack.[8] Even if there is no general base within the active site to deprotonate the primer hydroxyl, the lowered pka of the metal-bound hydroxyl favors the formation of the 3’-hydroxide nucleophile.[8] Metal ions and Lys522 contact non-bridging oxygens on the α-phosphate to stabilize the negative charge developing on the α-phosphorus during bond formation with the nucleophile.
Moreover, the Lys522 sidechain also moves to neutralize the negatively charged pyrophosphate group. Tyr526, His506, Arg518 side chains and the oxygen from the backbone carbonyl group of Ala476 take part in the hydrogen bond network and assist in aligning the substrate for phosphoryl transfer.[8]
Accessory proteins
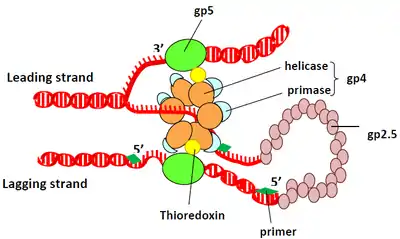
While phage T7 mediates DNA replication in very similar manner to higher organisms, T7 system is generally simpler compared to other replication systems. In addition to T7 DNA polymerase (also known as gp5), T7 replisome requires only four accessory proteins for proper function: host thioredoxin, gp4, gp2.5, and gp1.7.
Host thioredoxin
T7 polymerase by itself has a very low processivity. It dissociates from the primer-template after incorporating about 15 nucleotides. Upon infection of the host, T7 polymerase binds to host thioredoxin in 1:1 ratio. The hydrophobic interaction between thioredoxin and T7 polymerase helps to stabilize the binding of T7 polymerase to primer-template. In addition, the binding of thioredoxin increases T7 polymerase processivity to nearly 80-fold.[9] The precise mechanism for how the thioredoxin-T7 polymerase complex is able to achieve such increase in processivity is still unknown. Binding of thioredoxin exposes a large number of basic amino acid residues in the thumb region of T7 polymerase. Several studies suggest that the electrostatic interaction between these positively charged basic residues with the negatively charged phosphate backbone of DNA and other accessory proteins is responsible for increased processivity in gp5/thioredoxin complex.[9][10][11]
gp4
gp4 is a hexameric protein containing two functional domains: helicase domain and primase domain. The helicase domain unwinds double-stranded DNA to provide template for replication. The C-terminal tail of helicase domain contains several negatively charged acidic residues which make contact with the exposed basic residue of T7 polymerase/thioredoxin. These interactions help to load T7 polymerase/thioredoxin complex onto replication fork. The primase domain catalyzes the synthesis of short oligoribonucleotides. These oligoribonucleotides, called primers, are complementary to the template strand and used to initiate DNA replication. In T7 system, primase domain of one subunit interacts with primase domain of adjacent subunit. This interaction between primase domains acts as a brake to stop helicase when needed, which ensure the leading stand synthesis in-pace with lagging stand synthesis.[11]
gp2.5
| Single-stranded DNA-binding protein | |||||||
|---|---|---|---|---|---|---|---|
| Identifiers | |||||||
| Organism | |||||||
| Symbol | 2.5 | ||||||
| UniProt | P03696 | ||||||
| |||||||
gp2.5 has similar function to single-stranded DNA binding protein. gp2.5 protects single-stranded DNA produced during replication and coordinates synthesis of leading and lagging strands through interaction between its acidic C-terminal tail and gp5/thioredoxin.[11]
gp1.7
| Nucleotide kinase | |||||||
|---|---|---|---|---|---|---|---|
| Identifiers | |||||||
| Organism | |||||||
| Symbol | 1.7 | ||||||
| UniProt | P03781 | ||||||
| |||||||
gp1.7 is a nucleoside monophosphate kinase, which catalyzes the conversion of deoxynucleoside 5'-monophosphates to di and triphosphate nucleotides, which accounts for the sensitivity of T7 polymerase to dideoxynucleotides (see Sequenase below).[11]
Properties
Processivity
The primary gp5 subunit of T7 DNA Polymerase by itself has low processivity and dissociates from DNA after the incorporation of just a few nucleotides. In order to become efficiently processive, T7 DNA polymerase recruits host thioredoxin to form a thioredoxin-gp5 complex. Thioredoxin binds the thioredoxin binding domain of gp5 thereby stabilizes a flexible DNA binding region of gp5. The stabilization of this region of gp5 allosterically increases the amount of protein surface interaction with the duplex portion of the primer-template. The resulting thioredoxin-gp5 complex increases the affinity of T7 polymerase for the primer terminus by ~80-fold and acts processively around 800 nucleotide incorporation steps.[12]
The mechanism adopted by T7 polymerase to achieve its processivity differs from many other polymerases in that it does not rely on a DNA clamp or a clamp loader. Instead, the T7 DNA polymerase complex requires only three proteins for processive DNA polymerization: T7 polymerase (gp5), Escherichia coli thioredoxin, and single-stranded DNA-binding protein gp2.5.[13] Although these three proteins are the only ones required for template single-stranded DNA polymerization, in a native biological setting the thioredoxin-gp5 interacts with gp4 helicase, which provides single-stranded DNA template (figure 4). During leading strand synthesis thioredoxin-gp5 and gp4 form a high affinity complex increasing overall polymerase processivity to around 5 kb.[14][15]
Exonuclease activity
T7 DNA polymerase possesses a 3’-5’ single and double stranded DNA exonuclease activity. This exonuclease activity is activated when a newly synthesized base does not correctly base-pair with the template strand. Excision of incorrectly incorporated bases acts as a proofreading mechanism thereby increasing the fidelity of T7 polymerase.[4] During early characterization of exonuclease activity, it was discovered that iron-catalyzed oxidation of T7 polymerase produced a modified enzyme with greatly reduced exonuclease activity. This discovery lead to the development and use of T7 Polymerase as a sequenase in early DNA sequencing methods.[16]
The mechanism by which T7 DNA polymerase senses that a mismatched base has been incorporated is still a topic of study. However, some studies have provided evidence to suggesting that changes in tension of the template DNA strand caused by base-pair mismatch may induce exonuclease activation. Wuite et al. observed that applying tension of above 40 pN to the template DNA resulted in 100-fold increase in exonuclease activity.[17]
Applications
Strand extensions in site directed mutagenesis
Site-directed mutagenesis is a molecular biology method that is used to make specific and intentional changes to the DNA sequence of a gene and any gene products. The technique was developed at a time when the highest quality commercially available DNA polymerase for converting an oligonucleotide into a complete complementary DNA strand was the large (Klenow) fragment of E. coli DNA polymerase 1. However, ligation step can become an issue with oligonucleotide mutagenesis. That is when the DNA ligase operates inefficiently relative to the DNA polymerase, strand displacement of the oligonucleotide can reduce the mutant frequency. In the other hand, T7 DNA polymerase does not perform strand displacement synthesis; and thus, can be utilized to obtain high mutant frequencies for point mutants independent of ligation.[18]
Second strand synthesis of cDNA
cDNA cloning is a major technology for analysis of the expression of genomes. The full-length first-strand can be synthesized through the commercially available reverse transcriptases. Synthesis of the second-strand was once a major limitation to cDNA cloning. Two groups of methods differing by the mechanism of initiation were developed to synthesize the second-strand. In a first group of methods, initiation of second-strand synthesis takes place within the sequence of the first strand. However, the digestion of the 3' end of the first strand is required and therefore results in the loss of the sequences corresponding to the 5'end of the mRNA. In a second group of methods, initiation of second-strand synthesis takes place outside the sequence of the first strand. This group of methods does not require digestion of the 3' end of the first strand. However, the limitation of this group of method lies upon the elongation. Cloning with T7 DNA polymerase helps overcome this limitation by allowing digestion of the poly(dT) tract during the second-strand synthesis reaction. Therefore, the size of the tract synthesized with terminal transferase is not required to be within a given size range and the resulting clones contain a tract of a limited size. Moreover, due to high 3’ exonuclease activity of T7 DNA polymerase, high yield of the full-length second-strand can be obtained.[19]
Sequenase (DNA sequencing)
In Sanger sequencing, one of the major problem regarding DNA polymerases is the discrimination against dideoxynucleotides, the chain-terminating nucleotides. Most of known DNA polymerases strongly discriminate against ddNTP; and thus, a high ratio of ddNTP to dNTP must be used for efficient chain-termination. T7 DNA polymerase discriminates against ddNTP only several fold; and thereby, requires much lower concentration of ddNTP to provide high uniformity of DNA bands on the gel. However, its strong 3’-5’ exonuclease activity can disrupt the sequencing since when the concentration of dNTP falls, the exonuclease activity increases resulting in no net DNA synthesis or degradation of DNA. In order to use for DNA sequencing, T7 DNA polymerase has been modified to remove its exonuclease activity, either chemically (Sequenase 1.0) or by deletion of residues (Sequenase Version 2.0).[4][20]
References
- Mark DF, Richardson CC (March 1976). "Escherichia coli thioredoxin: a subunit of bacteriophage T7 DNA polymerase". Proceedings of the National Academy of Sciences of the United States of America. 73 (3): 780–4. Bibcode:1976PNAS...73..780M. doi:10.1073/pnas.73.3.780. PMC 336002. PMID 768986.
- Tabor S, Huber HE, Richardson CC (November 1987). "Escherichia coli thioredoxin confers processivity on the DNA polymerase activity of the gene 5 protein of bacteriophage T7". The Journal of Biological Chemistry. 262 (33): 16212–23. doi:10.1016/S0021-9258(18)47718-6. PMID 3316214.
- Venkitaraman AR (April 1989). "Use of modified T7 DNA polymerase (sequenase version 2.0) for oligonucleotide site-directed mutagenesis". Nucleic Acids Research. 17 (8): 3314. doi:10.1093/nar/17.8.3314. PMC 317753. PMID 2726477.
- Zhu B (16 April 2014). "Bacteriophage T7 DNA polymerase - sequenase". Frontiers in Microbiology. 5: 181. doi:10.3389/fmicb.2014.00181. PMC 3997047. PMID 24795710.
- "Thermo Sequenase DNA Polymerase".
- Voet D, Voet JG (2011). Biochemistry (4th ed.). Hoboken, NJ: John Wiley & Sons. ISBN 9780470917459.
- Fersht, Alan (1985). Enzyme structure and mechanism (2nd ed.). New York: W.H. Freeman. ISBN 9780716716143.
- Doublié S, Ellenberger T (December 1998). "The mechanism of action of T7 DNA polymerase". Current Opinion in Structural Biology. 8 (6): 704–12. doi:10.1016/s0959-440x(98)80089-4. PMID 9914251.
- Bedford E, Tabor S, Richardson CC (January 1997). "The thioredoxin binding domain of bacteriophage T7 DNA polymerase confers processivity on Escherichia coli DNA polymerase I". Proceedings of the National Academy of Sciences of the United States of America. 94 (2): 479–84. Bibcode:1997PNAS...94..479B. doi:10.1073/pnas.94.2.479. PMC 19538. PMID 9012809.
- Ghosh S, Hamdan SM, Cook TE, Richardson CC (November 2008). "Interactions of Escherichia coli thioredoxin, the processivity factor, with bacteriophage T7 DNA polymerase and helicase". The Journal of Biological Chemistry. 283 (46): 32077–84. doi:10.1074/jbc.M805062200. PMC 2581581. PMID 18757858.
- Lee SJ, Richardson CC (October 2011). "Choreography of bacteriophage T7 DNA replication". Current Opinion in Chemical Biology. 15 (5): 580–6. doi:10.1016/j.cbpa.2011.07.024. PMC 3195405. PMID 21907611.
- Richardson, CC (1983). "Bacteriophage T7: minimal requirements for the replication of a duplex DNA molecule". Cell. 33 (2): 315–317. doi:10.1016/0092-8674(83)90411-7. PMID 6344999.
- Kelman Z, Hurwitz J, O'Donnell M (February 1998). "Processivity of DNA polymerases: two mechanisms, one goal". Structure. 6 (2): 121–5. doi:10.1016/s0969-2126(98)00014-8. PMID 9519403.
- Akabayov B, Akabayov SR, Lee SJ, Tabor S, Kulczyk AW, Richardson CC (August 2010). "Conformational dynamics of bacteriophage T7 DNA polymerase and its processivity factor, Escherichia coli thioredoxin". Proceedings of the National Academy of Sciences of the United States of America. 107 (34): 15033–8. Bibcode:2010PNAS..10715033A. doi:10.1073/pnas.1010141107. PMC 2930546. PMID 20696935.
- Hamdan SM, Johnson DE, Tanner NA, Lee JB, Qimron U, Tabor S, van Oijen AM, Richardson CC (August 2007). "Dynamic DNA helicase-DNA polymerase interactions assure processive replication fork movement" (PDF). Molecular Cell. 27 (4): 539–49. doi:10.1016/j.molcel.2007.06.020. PMID 17707227.
- Tabor S, Richardson CC (November 1987). "Selective oxidation of the exonuclease domain of bacteriophage T7 DNA polymerase". The Journal of Biological Chemistry. 262 (32): 15330–3. doi:10.1016/S0021-9258(18)47726-5. PMID 2824455.
- Wuite GJ, Smith SB, Young M, Keller D, Bustamante C (March 2000). "Single-molecule studies of the effect of template tension on T7 DNA polymerase activity". Nature. 404 (6773): 103–6. Bibcode:2000Natur.404..103W. doi:10.1038/35003614. PMID 10716452. S2CID 2270107.
- Bebenek K, Kunkel TA (July 1989). "The use of native T7 DNA polymerase for site-directed mutagenesis". Nucleic Acids Research. 17 (13): 5408. doi:10.1093/nar/17.13.5408. PMC 318147. PMID 2668888.
- Bodescot M, Brison O (September 1994). "Efficient second-strand cDNA synthesis using T7 DNA polymerase". DNA and Cell Biology. 13 (9): 977–85. doi:10.1089/dna.1994.13.977. PMID 7522464.
- Fuller, CW; McArdle, BF; Griffin, AM; Griffin, HG (1996). DNA sequencing using sequenase version 2.0 T7 DNA polymerase. Methods in Molecular Biology. Vol. 58. pp. 373–87. doi:10.1385/0-89603-402-X:373. ISBN 0-89603-402-X. PMID 8713887.