Eukaryotic DNA replication
Eukaryotic DNA replication is a conserved mechanism that restricts DNA replication to once per cell cycle. Eukaryotic DNA replication of chromosomal DNA is central for the duplication of a cell and is necessary for the maintenance of the eukaryotic genome.
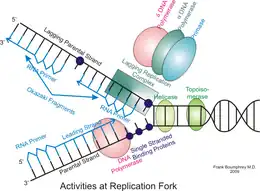
DNA replication is the action of DNA polymerases synthesizing a DNA strand complementary to the original template strand. To synthesize DNA, the double-stranded DNA is unwound by DNA helicases ahead of polymerases, forming a replication fork containing two single-stranded templates. Replication processes permit copying a single DNA double helix into two DNA helices, which are divided into the daughter cells at mitosis. The major enzymatic functions carried out at the replication fork are well conserved from prokaryotes to eukaryotes, but the replication machinery in eukaryotic DNA replication is a much larger complex, coordinating many proteins at the site of replication, forming the replisome.[1]
The replisome is responsible for copying the entirety of genomic DNA in each proliferative cell. This process allows for the high-fidelity passage of hereditary/genetic information from parental cell to daughter cell and is thus essential to all organisms. Much of the cell cycle is built around ensuring that DNA replication occurs without errors.[1]
In G1 phase of the cell cycle, many of the DNA replication regulatory processes are initiated. In eukaryotes, the vast majority of DNA synthesis occurs during S phase of the cell cycle, and the entire genome must be unwound and duplicated to form two daughter copies. During G2, any damaged DNA or replication errors are corrected. Finally, one copy of the genomes is segregated into each daughter cell at the mitosis or M phase.[2] These daughter copies each contains one strand from the parental duplex DNA and one nascent antiparallel strand.
This mechanism is conserved from prokaryotes to eukaryotes and is known as semiconservative DNA replication. The process of semiconservative replication for the site of DNA replication is a fork-like DNA structure, the replication fork, where the DNA helix is open, or unwound, exposing unpaired DNA nucleotides for recognition and base pairing for the incorporation of free nucleotides into double-stranded DNA.[3]
Initiation
Initiation of eukaryotic DNA replication is the first stage of DNA synthesis where the DNA double helix is unwound and an initial priming event by DNA polymerase α occurs on the leading strand. The priming event on the lagging strand establishes a replication fork. Priming of the DNA helix consists of the synthesis of an RNA primer to allow DNA synthesis by DNA polymerase α. Priming occurs once at the origin on the leading strand and at the start of each Okazaki fragment on the lagging strand.
Origin of replication
Replication starts at origins of replication. DNA sequences containing these sites were initially isolated in the late 1970s on the basis of their ability to support replication of plasmids, hence the designation of autonomously replicating sequences (ARS). Origins vary widely in their efficiency, with some being used in almost every cell cycle while others may be used in only one in one thousand S phases.[4] The total number of yeast ARSs is at least 1600, but may be more than 5000 if less active sites are counted,[5] that is, there may be an ARS every 2000 to 8000 base pairs.
Pre-replicative complex
Multiple replicative proteins assemble on and dissociate from these replicative origins to initiate DNA replication.[6] with the formation of the pre-replication complex (pre-RC) being a key intermediate in the replication initiation process.
Association of the origin recognition complex (ORC) with a replication origin recruits the cell division cycle 6 protein (Cdc6) to form a platform for the loading of the minichromosome maintenance (Mcm 2–7) complex proteins, facilitated by the chromatin licensing and DNA replication factor 1 protein (Cdt1). The ORC, Cdc6, and Cdt1 together are required for the stable association of the Mcm2-7 complex with replicative origins during the G1 phase of the cell cycle.[7]
Eukaryotic origins of replication control the formation of several protein complexes that lead to the assembly of two bidirectional DNA replication forks. These events are initiated by the formation of the pre-replication complex (pre-RC) at the origins of replication. This process takes place in the G1 stage of the cell cycle. The pre-RC formation involves the ordered assembly of many replication factors including the origin recognition complex (ORC), Cdc6 protein, Cdt1 protein, and minichromosome maintenance proteins (Mcm2-7).[8][9] Once the pre-RC is formed, activation of the complex is triggered by two kinases, cyclin-dependent kinase 2 (CDK) and Dbf4-dependent kinase (DDK) that help transition the pre-RC to the initiation complex before the initiation of DNA replication. This transition involves the ordered assembly of additional replication factors to unwind the DNA and accumulate the multiple eukaryotic DNA polymerases around the unwound DNA. Central to the question of how bidirectional replication forks are established at replication origins is the mechanism by which ORC recruits two head-to-head Mcm2-7 complexes to every replication origin to form the pre-replication complex.[10][11][12]
Origin recognition complex
The first step in the assembly of the pre-replication complex (pre-RC) is the binding of the origin recognition complex (ORC) to the replication origin. In late mitosis, the Cdc6 protein joins the bound ORC followed by binding the Cdt1-Mcm2-7 complex.[13] ORC, Cdc6, and Cdt1 are all required to load the six protein minichromosome maintenance (Mcm 2–7) complex onto the DNA. The ORC is a six-subunit, Orc1p-6, protein complex that selects the replicative origin sites on DNA for initiation of replication and ORC binding to chromatin is regulated through the cell cycle.[8][14] Generally, the function and size of the ORC subunits are conserved throughout many eukaryotic genomes with the difference being their diverged DNA binding sites.
The most widely studied origin recognition complex is that of Saccharomyces cerevisiae or yeast which is known to bind to the autonomously replicating sequence (ARS).[15] The S. cerevisiae ORC interacts specifically with both the A and B1 elements of yeast origins of replication, spanning a region of 30 base pairs.[16] The binding to these sequences requires ATP.[8][16]
The atomic structure of the S. cerevisiae ORC bound to ARS DNA has been determined.[16] Orc1, Orc2, Orc3, Orc4, and Orc5 encircle the A element by means of two types of interactions, base non-specific and base-specific, that bend the DNA at the A element. All five subunits contact the sugar phosphate backbone at multiple points of the A element to form a tight grip without base specificity. Orc1 and Orc2 contact the minor groove of the A element while a winged helix domain of Orc4 contacts the methyl groups of the invariant Ts in the major groove of the A element via an insertion helix (IH). The absence of this IH in metazoans[16] explains the lack of sequence specificity in human ORC.[17] Removing the IH from the ScORC causes it to lose its specificity for the A element, and to bind promiscuously and preferentially (83%) to promoter regions.[18] The ARS DNA is also bent at the B1 element through interactions with Orc2, Orc5 and Orc6.[16] The bending of origin DNA by ORC appears to be evolutionarily conserved suggesting that it may be required for the Mcm2-7 complex loading mechanism.[16][19]
When the ORC binds to DNA at replication origins, it serves as a scaffold for the assembly of other key initiation factors of the pre-replicative complex.[20] This pre-replicative complex assembly during the G1 stage of the cell cycle is required prior to the activation of DNA replication during the S phase.[21] The removal of at least part of the complex (Orc1) from the chromosome at metaphase is part of the regulation of mammalian ORC to ensure that the pre-replicative complex formation prior to the completion of metaphase is eliminated.[22]
Cdc6 protein
Binding of the cell division cycle 6 (Cdc6) protein to the origin recognition complex (ORC) is an essential step in the assembly of the pre-replication complex (pre-RC) at the origins of replication. Cdc6 binds to the ORC on DNA in an ATP-dependent manner, which induces a change in the pattern of origin binding that requires Orc1 ATPase.[23] Cdc6 requires ORC in order to associate with chromatin and is in turn required for the Cdt1-Mcm2-7 heptamer[13] to bind to the chromatin.[24] The ORC-Cdc6 complex forms a ring-shaped structure and is analogous to other ATP-dependent protein machines. The levels and activity of Cdc6 regulate the frequency with which the origins of replication are utilized during the cell cycle.
Cdt1 protein
The chromatin licensing and DNA replication factor 1 (Cdt1) protein is required for the licensing of chromatin for DNA replication.[25][26] In S. cerevisiae, Cdt1 facilitates the loading of the Mcm2-7 complex one at a time onto the chromosome by stabilising the left-handed open-ring structure of the Mcm2-7 single hexamer.[13][27][28] Cdt1 has been shown to associate with the C terminus of Cdc6 to cooperatively promote the association of Mcm proteins to the chromatin.[29] The cryo-EM structure of the OCCM (ORC-Cdc6-Cdt1-MCM) complex shows that the Cdt1-CTD interacts with the Mcm6-WHD.[30] In metazoans, Cdt1 activity during the cell cycle is tightly regulated by its association with the protein geminin, which both inhibits Cdt1 activity during S phase in order to prevent re-replication of DNA and prevents it from ubiquitination and subsequent proteolysis.[31]
Minichromosome maintenance protein complex
The minichromosome maintenance (Mcm) proteins were named after a genetic screen for DNA replication initiation mutants in S. cerevisiae that affect plasmid stability in an ARS-specific manner.[32] Mcm2, Mcm3, Mcm4, Mcm5, Mcm6 and Mcm7 form a hexameric complex that has an open-ring structure with a gap between Mcm2 and Mcm5.[13] The assembly of the Mcm proteins onto chromatin requires the coordinated function of the origin recognition complex (ORC), Cdc6, and Cdt1.[33] Once the Mcm proteins have been loaded onto the chromatin, ORC and Cdc6 can be removed from the chromatin without preventing subsequent DNA replication. This observation suggests that the primary role of the pre-replication complex is to correctly load the Mcm proteins.[34]
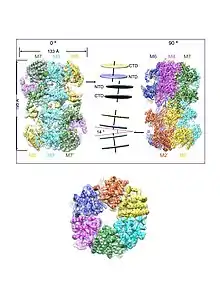
The Mcm proteins on chromatin form a head-to-head double hexamer with the two rings slightly tilted, twisted and off-centred to create a kink in the central channel where the bound DNA is captured at the interface of the two rings.[35][36] Each hexameric Mcm2-7 ring first serves as the scaffold for the assembly of the replisome and then as the core of the catalytic CMG (Cdc45-MCM-GINS) helicase, which is a main component of the replisome. Each Mcm protein is highly related to all others, but unique sequences distinguishing each of [18] the subunit types are conserved across eukaryotes. All eukaryotes have exactly six Mcm protein analogs that each fall into one of the existing classes (Mcm2-7), indicating that each Mcm protein has a unique and important function.[37][11]
Minichromosome maintenance proteins are required for DNA helicase activity. Inactivation of any of the six Mcm proteins during S phase irreversibly prevents further progression of the replication fork suggesting that the helicase cannot be recycled and must be assembled at replication origins.[38] Along with the minichromosome maintenance protein complex helicase activity, the complex also has associated ATPase activity.[39] Studies have shown that within the Mcm protein complex are specific catalytic pairs of Mcm proteins that function together to coordinate ATP hydrolysis.[40] These studies, confirmed by cryo-EM structures of the Mcm2-7 complexes,[13][35] showed that the Mcm complex is a hexamer with subunits arranged in a ring in the order of Mcm2-Mcm6-Mcm4-Mcm7-Mcm3-Mcm5-. Both members of each catalytic pair contribute to the conformation that allows ATP binding and hydrolysis and the mixture of active and inactive subunits presumably allows the Mcm hexameric complex to complete ATP binding and hydrolysis as a whole to create a coordinated ATPase activity.[41]
The nuclear localization of the minichromosome maintenance proteins is regulated in budding yeast cells.[42][43] The Mcm proteins are present in the nucleus in G1 stage and S phase of the cell cycle, but are exported to the cytoplasm during the G2 stage and M phase. A complete and intact six subunit Mcm complex is required to enter into the cell nucleus.[44] In S. cerevisiae, nuclear export is promoted by cyclin-dependent kinase (CDK) activity. Mcm proteins that are associated with chromatin are protected from CDK export machinery due to the lack of accessibility to CDK.[45]
Initiation complex
During the G1 stage of the cell cycle, the replication initiation factors, origin recognition complex (ORC), Cdc6, Cdt1, and minichromosome maintenance (Mcm) protein complex, bind sequentially to DNA to form a head-to-head dimer of the MCM ring complex, known as the pre-replication complex (pre-RC). While the yeast pre-RC forms a closed DNA complex,[35][36][46] the human pre-RC forms an open complex.[47] At the transition of the G1 stage to the S phase of the cell cycle, S phase–specific cyclin-dependent protein kinase (CDK) and Cdc7/Dbf4 kinase (DDK) transform the inert pre-RC into an active complex capable of assembling two bidirectional replisomes. CryoEM structures [48][49][50] showed that two DDKs independently dock onto the interface of the MCM double hexamer straddling across the two rings. The sequential phosphorylation of multiple substrates on the NTEs of Mcm4, Mcm2 and Mcm6 is achieved by a wobble mechanism whereby Dbf4 assumes different wobble states to position Cdc7 over its multiple substrates.[50] Phosphorylation of the MCM double hexamer, the Mcm4-NSD in particular, by DDK is essential for viability in yeast.[51] The recruitment of Cdc45 and GINS follows after the activation of the MCMs by DDK and CDK.
Cdc45 protein
Cell division cycle 45 (Cdc45) protein is a critical component for the conversion of the pre-replicative complex to the initiation complex. The Cdc45 protein assembles at replication origins before initiation and is required for replication to begin in Saccharomyces cerevisiae, and has an essential role during elongation. Thus, Cdc45 has central roles in both initiation and elongation phases of chromosomal DNA replication.[52]
Cdc45 associates with chromatin after the beginning of initiation in late G1 stage and during the S phase of the cell cycle. Cdc45 physically associates with Mcm5 and displays genetic interactions with five of the six members of the Mcm gene family and the ORC2 gene.[53][54] The loading of Cdc45 onto chromatin is critical for loading other various replication proteins, including DNA polymerase α, DNA polymerase ε, replication protein A (RPA) and proliferating cell nuclear antigen (PCNA) onto chromatin.[55][56][57][58] Within a Xenopus nucleus-free system, it has been demonstrated that Cdc45 is required for the unwinding of plasmid DNA.[58] The Xenopus nucleus-free system also demonstrates that DNA unwinding and tight RPA binding to chromatin occurs only in the presence of Cdc45.[55]
Binding of Cdc45 to chromatin depends on Clb-Cdc28 kinase activity as well as functional Cdc6 and Mcm2, which suggests that Cdc45 associates with the pre-RC after activation of S-phase cyclin-dependent kinases (CDKs). As indicated by the timing and the CDK dependence, binding of Cdc45 to chromatin is crucial for commitment to initiation of DNA replication. During S phase, Cdc45 physically interacts with Mcm proteins on chromatin; however, dissociation of Cdc45 from chromatin is slower than that of the Mcm, which indicates that the proteins are released by different mechanisms.[37]
GINS
The six minichromosome maintenance proteins and Cdc45 are essential during initiation and elongation for the movement of replication forks and for unwinding of the DNA. GINS are essential for the interaction of Mcm and Cdc45 at the origins of replication during initiation and then at DNA replication forks as the replisome progresses.[59][60] The GINS complex is composed of four small proteins Sld5 (Cdc105), Psf1 (Cdc101), Psf2 (Cdc102) and Psf3 (Cdc103), GINS represents 'go, ichi, ni, san' which means '5, 1, 2, 3' in Japanese.[61] Cdc45, Mcm2-7 and GINS together form the CMG helicase,[62] the replicative helicase of the replisome. Although the Mcm2-7 complex alone has weak helicase activity [63] Cdc45 and GINS are required for robust helicase activity[64][65]
Mcm10
Mcm10 is essential for chromosome replication and interacts with the minichromosome maintenance 2-7 helicase that is loaded in an inactive form at origins of DNA replication.[66][67] Mcm10 also chaperones the catalytic DNA polymerase α and helps stabilize the polymerase at replication forks.[68][69]
DDK and CDK kinases
At the onset of S phase, the pre-replicative complex must be activated by two S phase-specific kinases in order to form an initiation complex at an origin of replication. One kinase is the Cdc7-Dbf4 kinase called Dbf4-dependent kinase (DDK) and the other is cyclin-dependent kinase (CDK).[70] Chromatin-binding assays of Cdc45 in yeast and Xenopus have shown that a downstream event of CDK action is loading of Cdc45 onto chromatin.[71][72] Cdc6 has been speculated to be a target of CDK action, because of the association between Cdc6 and CDK, and the CDK-dependent phosphorylation of Cdc6. The CDK-dependent phosphorylation of Cdc6 has been considered to be required for entry into the S phase.[73]
Both the catalytic subunits of DDK, Cdc7, and the activator protein, Dbf4, are conserved in eukaryotes and are required for the onset of S phase of the cell cycle.[74][75] Both Dbf4 and Cdc7 are required for the loading of Cdc45 onto chromatin origins of replication. The target for binding of the DDK kinase is the chromatin-bound form of the Mcm complex.[76][77] High resolution cryoEM structures showed that the Dbf4 subunit of DDK straddles across the hexamer interface of the DNA-bound MCM-DH, contacting Mcm2 of one hexamer and Mcm4/6 of the opposite hexamer.[48][49][50] Mcm2, Mcm4 and Mcm6 are all substrates of phosphorylation by DDK [78][74] but only the N-terminal serine/threonine-rich domain (NSD) of Mcm4 is an essential DDK target.[50][51] Phosphorylation of the NSD leads to the activation of Mcm helicase activity.
Dpb11, Sld3, and Sld2 proteins
Sld3, Sld2, and Dpb11 interact with many replication proteins. Sld3 and Cdc45 form a complex that associated with the pre-RC at the early origins of replication even in the G11 phase and with the later origins of replication in the S phase in a mutually Mcm-dependent manner.[79][80] Dpb11 and Sld2 interact with Polymerase ɛ and cross-linking experiments have indicated that Dpb11 and Polymerase ɛ coprecipitate in the S phase and associate with replication origins.[81][82]
Sld3 and Sld2 are phosphorylated by CDK, which enables the two replicative proteins to bind to Dpb11. Dpb11 had two pairs of BRCA1 C Terminus (BRCT) domains which are known as a phosphopeptide-binding domains.[83] The N-terminal pair of the BRCT domains binds to phosphorylated Sld3, and the C-terminal pair binds to phosphorylated Sld2. Both of these interactions are essential for CDK-dependent activation of DNA budding in yeast.[84]
Dpb11 also interacts with GINS and participates in the initiation and elongation steps of chromosomal DNA replication.[60][85][86] GINS are one of the replication proteins found at the replication forks and forms a complex with Cdc45 and Mcm.
These phosphorylation-dependent interactions between Dpb11, Sld2, and Sld3 are essential for CDK-dependent activation of DNA replication, and by using cross-linking reagents within some experiments, a fragile complex was identified called the pre-loading complex (pre-LC). This complex contains Pol ɛ, GINS, Sld2, and Dpb11. The pre-LC is found to form before any association with the origins in a CDK-dependent and DDK-dependent manner and CDK activity regulates the initiation of DNA replication through the formation of the pre-LC.[87]
Elongation
The formation of the pre-replicative complex (pre-RC) marks the potential sites for the initiation of DNA replication. Consistent with the minichromosome maintenance complex encircling double stranded DNA, formation of the pre-RC does not lead to the immediate unwinding of origin DNA or the recruitment of DNA polymerases. Instead, the pre-RC that is formed during the G1 of the cell cycle is only activated to unwind the DNA and initiate replication after the cells pass from the G1 to the S phase of the cell cycle.[2]
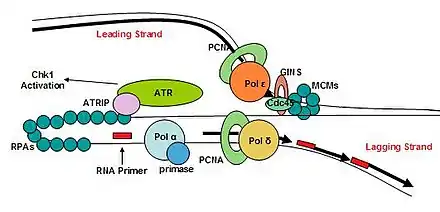
Once the initiation complex is formed and the cells pass into the S phase, the complex then becomes a replisome. The eukaryotic replisome complex is responsible for coordinating DNA replication. Replication on the leading and lagging strands is performed by DNA polymerase ε and DNA polymerase δ. Many replisome factors including Claspin, And1, replication factor C clamp loader and the fork protection complex are responsible for regulating polymerase functions and coordinating DNA synthesis with the unwinding of the template strand by Cdc45-Mcm-GINS complex. As the DNA is unwound the twist number decreases. To compensate for this the writhe number increases, introducing positive supercoils in the DNA. These supercoils would cause DNA replication to halt if they were not removed. Topoisomerases are responsible for removing these supercoils ahead of the replication fork.
The replisome is responsible for copying the entire genomic DNA in each proliferative cell. The base pairing and chain formation reactions, which form the daughter helix, are catalyzed by DNA polymerases.[88] These enzymes move along single-stranded DNA and allow for the extension of the nascent DNA strand by "reading" the template strand and allowing for incorporation of the proper purine nucleobases, adenine and guanine, and pyrimidine nucleobases, thymine and cytosine. Activated free deoxyribonucleotides exist in the cell as deoxyribonucleotide triphosphates (dNTPs). These free nucleotides are added to an exposed 3'-hydroxyl group on the last incorporated nucleotide. In this reaction, a pyrophosphate is released from the free dNTP, generating energy for the polymerization reaction and exposing the 5' monophosphate, which is then covalently bonded to the 3' oxygen. Additionally, incorrectly inserted nucleotides can be removed and replaced by the correct nucleotides in an energetically favorable reaction. This property is vital to proper proofreading and repair of errors that occur during DNA replication.
Replication fork
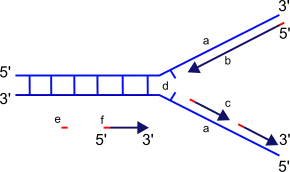
The replication fork is the junction between the newly separated template strands, known as the leading and lagging strands, and the double stranded DNA. Since duplex DNA is antiparallel, DNA replication occurs in opposite directions between the two new strands at the replication fork, but all DNA polymerases synthesize DNA in the 5' to 3' direction with respect to the newly synthesized strand. Further coordination is required during DNA replication. Two replicative polymerases synthesize DNA in opposite orientations. Polymerase ε synthesizes DNA on the "leading" DNA strand continuously as it is pointing in the same direction as DNA unwinding by the replisome. In contrast, polymerase δ synthesizes DNA on the "lagging" strand, which is the opposite DNA template strand, in a fragmented or discontinuous manner.
The discontinuous stretches of DNA replication products on the lagging strand are known as Okazaki fragments and are about 100 to 200 bases in length at eukaryotic replication forks. The lagging strand usually contains longer stretches of single-stranded DNA that is coated with single-stranded binding proteins, which help stabilize the single-stranded templates by preventing a secondary structure formation. In eukaryotes, these single-stranded binding proteins are a heterotrimeric complex known as replication protein A (RPA).[89]
Each Okazaki fragment is preceded by an RNA primer, which is displaced by the procession of the next Okazaki fragment during synthesis. RNase H recognizes the DNA:RNA hybrids that are created by the use of RNA primers and is responsible for removing these from the replicated strand, leaving behind a primer:template junction. DNA polymerase α, recognizes these sites and elongates the breaks left by primer removal. In eukaryotic cells, a small amount of the DNA segment immediately upstream of the RNA primer is also displaced, creating a flap structure. This flap is then cleaved by endonucleases. At the replication fork, the gap in DNA after removal of the flap is sealed by DNA ligase I, which repairs the nicks that are left between the 3'-OH and 5'phosphate of the newly synthesized strand.[90] Owing to the relatively short nature of the eukaryotic Okazaki fragment, DNA replication synthesis occurring discontinuously on the lagging strand is less efficient and more time-consuming than leading-strand synthesis. DNA synthesis is complete once all RNA primers are removed and nicks are repaired.
Leading strand
During DNA replication, the replisome will unwind the parental duplex DNA into a two single-stranded DNA template replication fork in a 5' to 3' direction. The leading strand is the template strand that is being replicated in the same direction as the movement of the replication fork. This allows the newly synthesized strand complementary to the original strand to be synthesized 5' to 3' in the same direction as the movement of the replication fork.[91]
Once an RNA primer has been added by a primase to the 3' end of the leading strand, DNA synthesis will continue in a 3' to 5' direction with respect to the leading strand uninterrupted. DNA Polymerase ε will continuously add nucleotides to the template strand therefore making leading strand synthesis require only one primer and has uninterrupted DNA polymerase activity.[92]
Lagging strand
DNA replication on the lagging strand is discontinuous. In lagging strand synthesis, the movement of DNA polymerase in the opposite direction of the replication fork requires the use of multiple RNA primers. DNA polymerase will synthesize short fragments of DNA called Okazaki fragments which are added to the 3' end of the primer. These fragments can be anywhere between 100 and 400 nucleotides long in eukaryotes.[93]
At the end of Okazaki fragment synthesis, DNA polymerase δ runs into the previous Okazaki fragment and displaces its 5' end containing the RNA primer and a small segment of DNA. This generates an RNA-DNA single strand flap, which must be cleaved, and the nick between the two Okazaki fragments must be sealed by DNA ligase I. This process is known as Okazaki fragment maturation and can be handled in two ways: one mechanism processes short flaps, while the other deals with long flaps.[94] DNA polymerase δ is able to displace up to 2 to 3 nucleotides of DNA or RNA ahead of its polymerization, generating a short "flap" substrate for Fen1, which can remove nucleotides from the flap, one nucleotide at a time.
By repeating cycles of this process, DNA polymerase δ and Fen1 can coordinate the removal of RNA primers and leave a DNA nick at the lagging strand.[95] It has been proposed that this iterative process is preferable to the cell because it is tightly regulated and does not generate large flaps that need to be excised.[96] In the event of deregulated Fen1/DNA polymerase δ activity, the cell uses an alternative mechanism to generate and process long flaps by using Dna2, which has both helicase and nuclease activities.[97] The nuclease activity of Dna2 is required for removing these long flaps, leaving a shorter flap to be processed by Fen1. Electron microscopy studies indicate that nucleosome loading on the lagging strand occurs very close to the site of synthesis.[98] Thus, Okazaki fragment maturation is an efficient process that occurs immediately after the nascent DNA is synthesized.
Replicative DNA polymerases
After the replicative helicase has unwound the parental DNA duplex, exposing two single-stranded DNA templates, replicative polymerases are needed to generate two copies of the parental genome. DNA polymerase function is highly specialized and accomplish replication on specific templates and in narrow localizations. At the eukaryotic replication fork, there are three distinct replicative polymerase complexes that contribute to DNA replication: Polymerase α, Polymerase δ, and Polymerase ε. These three polymerases are essential for viability of the cell.[99]
Because DNA polymerases require a primer on which to begin DNA synthesis, polymerase α (Pol α) acts as a replicative primase. Pol α is associated with an RNA primase and this complex accomplishes the priming task by synthesizing a primer that contains a short 10 nucleotide stretch of RNA followed by 10 to 20 DNA bases.[3] Importantly, this priming action occurs at replication initiation at origins to begin leading-strand synthesis and also at the 5' end of each Okazaki fragment on the lagging strand.
However, Pol α is not able to continue DNA replication and must be replaced with another polymerase to continue DNA synthesis.[100] Polymerase switching requires clamp loaders and it has been proven that normal DNA replication requires the coordinated actions of all three DNA polymerases: Pol α for priming synthesis, Pol ε for leading-strand replication, and the Pol δ, which is constantly loaded, for generating Okazaki fragments during lagging-strand synthesis.[101]
- Polymerase α (Pol α): Forms a complex with a small catalytic subunit (PriS) and a large noncatalytic (PriL) subunit.[102] First, synthesis of an RNA primer allows DNA synthesis by DNA polymerase alpha. Occurs once at the origin on the leading strand and at the start of each Okazaki fragment on the lagging strand. Pri subunits act as a primase, synthesizing an RNA primer. DNA Pol α elongates the newly formed primer with DNA nucleotides. After around 20 nucleotides, elongation is taken over by Pol ε on the leading strand and Pol δ on the lagging strand.[103]
- Polymerase δ (Pol δ): Highly processive and has proofreading, 3'->5' exonuclease activity. In vivo, it is the main polymerase involved in both lagging strand and leading strand synthesis.[104]
- Polymerase ε (Pol ε): Highly processive and has proofreading, 3'->5' exonuclease activity. Highly related to pol δ, in vivo it functions mainly in error checking of pol δ.[104]
Cdc45–Mcm–GINS helicase complex
The DNA helicases and polymerases must remain in close contact at the replication fork. If unwinding occurs too far in advance of synthesis, large tracts of single-stranded DNA are exposed. This can activate DNA damage signaling or induce DNA repair processes. To thwart these problems, the eukaryotic replisome contains specialized proteins that are designed to regulate the helicase activity ahead of the replication fork. These proteins also provide docking sites for physical interaction between helicases and polymerases, thereby ensuring that duplex unwinding is coupled with DNA synthesis.[105]
For DNA polymerases to function, the double-stranded DNA helix has to be unwound to expose two single-stranded DNA templates for replication. DNA helicases are responsible for unwinding the double-stranded DNA during chromosome replication. Helicases in eukaryotic cells are remarkably complex.[106] The catalytic core of the helicase is composed of six minichromosome maintenance (Mcm2-7) proteins, forming a hexameric ring. Away from DNA, the Mcm2-7 proteins form a single heterohexamer and are loaded in an inactive form at origins of DNA replication as a head-to-head double hexamers around double-stranded DNA.[106][107] The Mcm proteins are recruited to replication origins then redistributed throughout the genomic DNA during S phase, indicative of their localization to the replication fork.[54]
Loading of Mcm proteins can only occur during the G1 of the cell cycle, and the loaded complex is then activated during S phase by recruitment of the Cdc45 protein and the GINS complex to form the active Cdc45–Mcm–GINS (CMG) helicase at DNA replication forks.[62][108] Mcm activity is required throughout the S phase for DNA replication.[38][109] A variety of regulatory factors assemble around the CMG helicase to produce the ‘Replisome Progression Complex’ which associates with DNA polymerases to form the eukaryotic replisome, the structure of which is still quite poorly defined in comparison with its bacterial counterpart.[59][110]
The isolated CMG helicase and Replisome Progression Complex contain a single Mcm protein ring complex suggesting that the loaded double hexamer of the Mcm proteins at origins might be broken into two single hexameric rings as part of the initiation process, with each Mcm protein complex ring forming the core of a CMG helicase at the two replication forks established from each origin.[59][62] The full CMG complex is required for DNA unwinding, and the complex of CDC45-Mcm-GINS is the functional DNA helicase in eukaryotic cells.[64]
Ctf4 and And1 proteins
The CMG complex interacts with the replisome through the interaction with Ctf4 and And1 proteins. Ctf4/And1 proteins interact with both the CMG complex and DNA polymerase α.[111] Ctf4 is a polymerase α accessory factor, which is required for the recruitment of polymerase α to replication origins.[112]
Mrc1 and Claspin proteins
Mrc1/Claspin proteins couple leading-strand synthesis with the CMG complex helicase activity. Mrc1 interacts with polymerase ε as well as Mcm proteins.[113] The importance of this direct link between the helicase and the leading-strand polymerase is underscored by results in cultured human cells, where Mrc1/Claspin is required for efficient replication fork progression.[114] These results suggest that efficient DNA replication also requires the coupling of helicases and leading-strand synthesis...
Proliferating cell nuclear antigen
DNA polymerases require additional factors to support DNA replication. DNA polymerases have a semiclosed 'hand' structure, which allows the polymerase to load onto the DNA and begin translocating. This structure permits DNA polymerase to hold the single-stranded DNA template, incorporate dNTPs at the active site, and release the newly formed double-stranded DNA. However, the structure of DNA polymerases does not allow a continuous stable interaction with the template DNA.[1]
To strengthen the interaction between the polymerase and the template DNA, DNA sliding clamps associate with the polymerase to promote the processivity of the replicative polymerase. In eukaryotes, the sliding clamp is a homotrimer ring structure known as the proliferating cell nuclear antigen (PCNA). The PCNA ring has polarity with surfaces that interact with DNA polymerases and tethers them securely to the DNA template. PCNA-dependent stabilization of DNA polymerases has a significant effect on DNA replication because PCNAs are able to enhance the polymerase processivity up to 1,000-fold.[115][116] PCNA is an essential cofactor and has the distinction of being one of the most common interaction platforms in the replisome to accommodate multiple processes at the replication fork, and so PCNA is also viewed as a regulatory cofactor for DNA polymerases.[117]
Replication factor C
PCNA fully encircles the DNA template strand and must be loaded onto DNA at the replication fork. At the leading strand, loading of the PCNA is an infrequent process, because DNA replication on the leading strand is continuous until replication is terminated. However, at the lagging strand, DNA polymerase δ needs to be continually loaded at the start of each Okazaki fragment. This constant initiation of Okazaki fragment synthesis requires repeated PCNA loading for efficient DNA replication.
PCNA loading is accomplished by the replication factor C (RFC) complex. The RFC complex is composed of five ATPases: Rfc1, Rfc2, Rfc3, Rfc4 and Rfc5.[118] RFC recognizes primer-template junctions and loads PCNA at these sites.[119][120] The PCNA homotrimer is opened by RFC by ATP hydrolysis and is then loaded onto DNA in the proper orientation to facilitate its association with the polymerase.[121][122] Clamp loaders can also unload PCNA from DNA; a mechanism needed when replication must be terminated.[122]
Stalled replication fork
DNA replication at the replication fork can be halted by a shortage of deoxynucleotide triphosphates (dNTPs) or by DNA damage, resulting in replication stress.[123] This halting of replication is described as a stalled replication fork. A fork protection complex of proteins stabilizes the replication fork until DNA damage or other replication problems can be fixed.[123] Prolonged replication fork stalling can lead to further DNA damage. Stalling signals are deactivated if the problems causing the replication fork are resolved.[123]
Termination
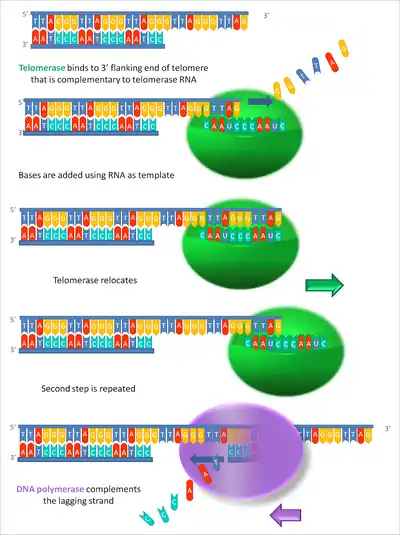
Termination of eukaryotic DNA replication requires different processes depending on whether the chromosomes are circular or linear. Unlike linear molecules, circular chromosomes are able to replicate the entire molecule. However, the two DNA molecules will remain linked together. This issue is handled by decatenation of the two DNA molecules by a type II topoisomerase. Type II topoisomerases are also used to separate linear strands as they are intricately folded into a nucleosome within the cell.
As previously mentioned, linear chromosomes face another issue that is not seen in circular DNA replication. Due to the fact that an RNA primer is required for initiation of DNA synthesis, the lagging strand is at a disadvantage in replicating the entire chromosome. While the leading strand can use a single RNA primer to extend the 5' terminus of the replicating DNA strand, multiple RNA primers are responsible for lagging strand synthesis, creating Okazaki fragments. This leads to an issue due to the fact that DNA polymerase is only able to add to the 3' end of the DNA strand. The 3'-5' action of DNA polymerase along the parent strand leaves a short single-stranded DNA (ssDNA) region at the 3' end of the parent strand when the Okazaki fragments have been repaired. Since replication occurs in opposite directions at opposite ends of parent chromosomes, each strand is a lagging strand at one end. Over time this would result in progressive shortening of both daughter chromosomes. This is known as the end replication problem.[1]
The end replication problem is handled in eukaryotic cells by telomere regions and telomerase. Telomeres extend the 3' end of the parental chromosome beyond the 5' end of the daughter strand. This single-stranded DNA structure can act as an origin of replication that recruits telomerase. Telomerase is a specialized DNA polymerase that consists of multiple protein subunits and an RNA component. The RNA component of telomerase anneals to the single stranded 3' end of the template DNA and contains 1.5 copies of the telomeric sequence.[93] Telomerase contains a protein subunit that is a reverse transcriptase called telomerase reverse transcriptase or TERT. TERT synthesizes DNA until the end of the template telomerase RNA and then disengages.[93] This process can be repeated as many times as needed with the extension of the 3' end of the parental DNA molecule. This 3' addition provides a template for extension of the 5' end of the daughter strand by lagging strand DNA synthesis. Regulation of telomerase activity is handled by telomere-binding proteins.
Replication fork barriers
Prokaryotic DNA replication is bidirectional; within a replicative origin, replisome complexes are created at each end of the replication origin and replisomes move away from each other from the initial starting point. In prokaryotes, bidirectional replication initiates at one replicative origin on the circular chromosome and terminates at a site opposed from the initial start of the origin.[124] These termination regions have DNA sequences known as Ter sites. These Ter sites are bound by the Tus protein. The Ter-Tus complex is able to stop helicase activity, terminating replication.[125]
In eukaryotic cells, termination of replication usually occurs through the collision of the two replicative forks between two active replication origins. The location of the collision varies on the timing of origin firing. In this way, if a replication fork becomes stalled or collapses at a certain site, replication of the site can be rescued when a replisome traveling in the opposite direction completes copying the region. There are programmed replication fork barriers (RFBs) bound by RFB proteins in various locations, throughout the genome, which are able to terminate or pause replication forks, stopping progression of the replisome.[124]
Replication factories
It has been found that replication happens in a localised way in the cell nucleus. Contrary to the traditional view of moving replication forks along stagnant DNA, a concept of replication factories emerged, which means replication forks are concentrated towards some immobilised 'factory' regions through which the template DNA strands pass like conveyor belts.[126]
Cell cycle regulation

DNA replication is a tightly orchestrated process that is controlled within the context of the cell cycle. Progress through the cell cycle and in turn DNA replication is tightly regulated by the formation and activation of pre-replicative complexes (pre-RCs) which is achieved through the activation and inactivation of cyclin-dependent kinases (Cdks, CDKs). Specifically it is the interactions of cyclins and cyclin dependent kinases that are responsible for the transition from G1 into S-phase.
During the G1 phase of the cell cycle there are low levels of CDK activity. This low level of CDK activity allows for the formation of new pre-RC complexes but is not sufficient for DNA replication to be initiated by the newly formed pre-RCs. During the remaining phases of the cell cycle there are elevated levels of CDK activity. This high level of CDK activity is responsible for initiating DNA replication as well as inhibiting new pre-RC complex formation.[2] Once DNA replication has been initiated the pre-RC complex is broken down. Due to the fact that CDK levels remain high during the S phase, G2, and M phases of the cell cycle no new pre-RC complexes can be formed. This all helps to ensure that no initiation can occur until the cell division is complete.
In addition to cyclin dependent kinases a new round of replication is thought to be prevented through the downregulation of Cdt1. This is achieved via degradation of Cdt1 as well as through the inhibitory actions of a protein known as geminin. Geminin binds tightly to Cdt1 and is thought to be the major inhibitor of re-replication.[2] Geminin first appears in S-phase and is degraded at the metaphase-anaphase transition, possibly through ubiquination by anaphase promoting complex (APC).[127]
Various cell cycle checkpoints are present throughout the course of the cell cycle that determine whether a cell will progress through division entirely. Importantly in replication the G1, or restriction, checkpoint makes the determination of whether or not initiation of replication will begin or whether the cell will be placed in a resting stage known as G0. Cells in the G0 stage of the cell cycle are prevented from initiating a round of replication because the minichromosome maintenance proteins are not expressed. Transition into the S-phase indicates replication has begun.
Replication checkpoint proteins
In order to preserve genetic information during cell division, DNA replication must be completed with high fidelity. In order to achieve this task, eukaryotic cells have proteins in place during certain points in the replication process that are able to detect any errors during DNA replication and are able to preserve genomic integrity. These checkpoint proteins are able to stop the cell cycle from entering mitosis in order to allow time for DNA repair. Checkpoint proteins are also involved in some DNA repair pathways, while they stabilize the structure of the replication fork to prevent further damage. These checkpoint proteins are essential to avoid passing down mutations or other chromosomal aberrations to offspring.
Eukaryotic checkpoint proteins are well conserved and involve two phosphatidylinositol 3-kinase-related kinases (PIKKs), ATR and ATM. Both ATR and ATM share a target phosphorylation sequence, the SQ/TQ motif, but their individual roles in cells differ.[128]
ATR is involved in arresting the cell cycle in response to DNA double-stranded breaks. ATR has an obligate checkpoint partner, ATR-interacting-protein (ATRIP), and together these two proteins are responsive to stretches of single-stranded DNA that are coated by replication protein A (RPA).[129] The formation of single-stranded DNA occurs frequently, more often during replication stress. ATR-ATRIP is able to arrest the cell cycle to preserve genome integrity. ATR is found on chromatin during S phase, similar to RPA and claspin.[130]
The generation of single-stranded DNA tracts is important in initiating the checkpoint pathways downstream of replication damage. Once single-stranded DNA becomes sufficiently long, single-stranded DNA coated with RPA are able to recruit ATR-ATRIP.[131] In order to become fully active, the ATR kinase rely on sensor proteins that sense whether the checkpoint proteins are localized to a valid site of DNA replication stress. The RAD9-HUS1-Rad1 (9-1-1) heterotrimeric clamp and its clamp loader RFCRad17 are able to recognize gapped or nicked DNA. The RFCRad17 clamp loader loads 9-1-1 onto the damaged DNA.[132] The presence of 9-1-1 on DNA is enough to facilitate the interaction between ATR-ATRIP and a group of proteins termed checkpoint mediators, such as TOPBP1 and Mrc1/claspin. TOPBP1 interacts with and recruits the phosphorylated Rad9 component of 9-1-1 and binds ATR-ATRIP, which phosphorylates Chk1.[133] Mrc1/Claspin is also required for the complete activation of ATR-ATRIP that phosphorylates Chk1, the major downstream checkpoint effector kinase.[134] Claspin is a component of the replisome and contains a domain for docking with Chk1, revealing a specific function of Claspin during DNA replication: the promotion of checkpoint signaling at the replisome.[135]
Chk1 signaling is vital for arresting the cell cycle and preventing cells from entering mitosis with incomplete DNA replication or DNA damage. The Chk1-dependent Cdk inhibition is important for the function of the ATR-Chk1 checkpoint and to arrest the cell cycle and allow sufficient time for completion of DNA repair mechanisms, which in turn prevents the inheritance of damaged DNA. In addition, Chk1-dependent Cdk inhibition plays a critical role in inhibiting origin firing during S phase. This mechanism prevents continued DNA synthesis and is required for the protection of the genome in the presence of replication stress and potential genotoxic conditions.[136] Thus, ATR-Chk1 activity further prevents potential replication problems at the level of single replication origins by inhibiting initiation of replication throughout the genome, until the signaling cascade maintaining cell-cycle arrest is turned off.
Replication through nucleosomes
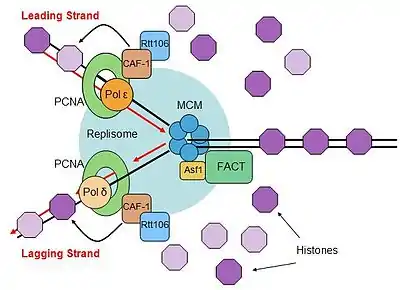
Eukaryotic DNA must be tightly compacted in order to fit within the confined space of the nucleus. Chromosomes are packaged by wrapping 147 nucleotides around an octamer of histone proteins, forming a nucleosome. The nucleosome octamer includes two copies of each histone H2A, H2B, H3, and H4. Due to the tight association of histone proteins to DNA, eukaryotic cells have proteins that are designed to remodel histones ahead of the replication fork, in order to allow smooth progression of the replisome.[137] There are also proteins involved in reassembling histones behind the replication fork to reestablish the nucleosome conformation.[138]
There are several histone chaperones that are known to be involved in nucleosome assembly after replication. The FACT complex has been found to interact with DNA polymerase α-primase complex, and the subunits of the FACT complex interacted genetically with replication factors.[139][140] The FACT complex is a heterodimer that does not hydrolyze ATP, but is able to facilitate "loosening" of histones in nucleosomes, but how the FACT complex is able to relieve the tight association of histones for DNA removal remains unanswered.[141]
Another histone chaperone that associates with the replisome is Asf1, which interacts with the Mcm complex dependent on histone dimers H3-H4.[142] Asf1 is able to pass newly synthesized H3-H4 dimer to deposition factors behind the replication fork and this activity makes the H3-H4 histone dimers available at the site of histone deposition just after replication.[143] Asf1 (and its partner Rtt109) has also been implicated in inhibiting gene expression from replicated genes during S-phase.[144]
The heterotrimeric chaperone chromatin assembly factor 1 (CAF-1) is a chromatin formation protein that is involved in depositing histones onto both newly replicated DNA strands to form chromatin.[145] CAF-1 contains a PCNA-binding motif, called a PIP-box, that allows CAF-1 to associate with the replisome through PCNA and is able to deposit histone H3-H4 dimers onto newly synthesized DNA.[146][147] The Rtt106 chaperone is also involved in this process, and associated with CAF-1 and H3-H4 dimers during chromatin formation.[148] These processes load newly synthesized histones onto DNA.
After the deposition of histones H3-H4, nucleosomes form by the association of histone H2A-H2B. This process is thought to occur through the FACT complex, since it already associated with the replisome and is able to bind free H2A-H2B, or there is the possibility of another H2A-H2B chaperone, Nap1.[149] Electron microscopy studies show that this occurs very quickly, as nucleosomes can be observed forming just a few hundred base pairs after the replication fork.[150] Therefore, the entire process of forming new nucleosomes takes place just after replication due to the coupling of histone chaperones to the replisome.
Comparisons between prokaryotic and eukaryotic DNA replication
When compared to prokaryotic DNA replication, the completion of eukaryotic DNA replication is more complex and involves multiple origins of replication and replicative proteins to accomplish. Prokaryotic DNA is arranged in a circular shape, and has only one replication origin when replication starts. By contrast, eukaryotic DNA is linear. When replicated, there are as many as one thousand origins of replication.[151]
Eukaryotic DNA is bidirectional. Here the meaning of the word bidirectional is different. Eukaryotic linear DNA has many origins (called O) and termini (called T). "T" is present to the right of "O". One "O" and one "T" together form one replicon. After the formation of pre-initiation complex, when one replicon starts elongation, initiation starts in second replicon. Now, if the first replicon moves in clockwise direction, the second replicon moves in anticlockwise direction, until "T" of first replicon is reached. At "T", both the replicons merge to complete the process of replication. Meanwhile, the second replicon is moving in forward direction also, to meet with the third replicon. This clockwise and counter-clockwise movement of two replicons is termed as bidirectional replication.
Eukaryotic DNA replication requires precise coordination of all DNA polymerases and associated proteins to replicate the entire genome each time a cell divides. This process is achieved through a series of steps of protein assemblies at origins of replication, mainly focusing the regulation of DNA replication on the association of the MCM helicase with the DNA. These origins of replication direct the number of protein complexes that will form to initiate replication. In prokaryotic DNA replication regulation focuses on the binding of the DnaA initiator protein to the DNA, with initiation of replication occurring multiple times during one cell cycle.[93] Both prokaryotic and eukaryotic DNA use ATP binding and hydrolysis to direct helicase loading and in both cases the helicase is loaded in the inactive form. However, eukaryotic helicases are double hexamers that are loaded onto double stranded DNA whereas prokaryotic helicases are single hexamers loaded onto single stranded DNA.[152]
Segregation of chromosomes is another difference between prokaryotic and eukaryotic cells. Rapidly dividing cells, such as bacteria, will often begin to segregate chromosomes that are still in the process of replication. In eukaryotic cells chromosome segregation into the daughter cells is not initiated until replication is complete in all chromosomes.[93] Despite these differences, however, the underlying process of replication is similar for both prokaryotic and eukaryotic DNA.
| Prokaryotic DNA replication | Eukaryotic DNA replication |
|---|---|
| Occurs inside the cytoplasm | Occurs inside the nucleus |
| Only one origin of replication per molecule of DNA | Have many origins of replication in each chromosome |
| Origin of replication is about 100-200 or more nucleotides in length | Each origin of replication is formed of about 150 nucleotides |
| Replication occurs at one point in each chromosome | Replication occurs at several points simultaneously in each chromosome |
| Only have one origin of replication | Has multiple origins of replication |
| Initiation is carried out by protein DnaA and DnaB | Initiation is carried out by the Origin Recognition Complex |
| Topoisomerase is needed | Topoisomerase is needed |
| Replication is very rapid | Replication is very slow |
Eukaryotic DNA replication protein list
List of major proteins involved in eukaryotic DNA replication:
| Protein | Function in Eukaryotic DNA replication |
|---|---|
| AND1 | Loads DNA polymerase α onto chromatin together with CMG complex on the lagging strand. Also known as Ctf4 in budding yeast. |
| Cdc45 | Required for initiation and elongation steps of DNA replication. A part of the Mcm2-7 helicase complex. Required after pre-RC step for loading of various proteins for initiation and elongation. |
| Cdc45-Mcm-GINS (CMG) complex | Functional DNA helicase in eukaryotic cells |
| Cdc6 | Required for assembly of Mcm2-7 complex at ORC, in conjunction with Cdt1 . |
| Cdc7-Dbf4 kinase or Dbf4-dependent kinase (DDK) | Protein kinase required for initiation of DNA replication, probably through phosphorylation of the minichromosome maintenance proteins. |
| Cdt1 | Loads Mcm2-7 complex on DNA at ORC in pre-RC/licensing step. Inhibited in metazoans by geminin. |
| Claspin | Couple leading-strand synthesis with the CMG complex helicase activity. Works with Mrc1 |
| Ctf4 | Loads DNA polymerase α onto chromatin together with CMG complex on the lagging strand. Homolog in metazoans is known as AND-1. |
| Cyclin-dependent kinase (CDK) | Cyclin-dependent protein kinase required for initiation of replication and for other subsequent steps. |
| Dna2 | 5' flap endonuclease and helicase involved in processing Okazaki fragments. |
| DNA ligase I | Joins Okazaki fragments during DNA replication. Ligase activity also needed for DNA repair and recombination. |
| DNA polymerase α (Pol α) | Contains primase activity that is necessary to initiate DNA synthesis on both leading and lagging strands. |
| DNA polymerase δ (Pol δ) | Required to complete synthesis of Okazaki fragments on the lagging strand that have been started by DNA polymerase α. |
| DNA polymerase ε (Pol ε) | The leading strand polymerase. Synthesizes DNA at the replication fork. Binds early at origins via Dbp11 and needed to load DNA polymerase α. |
| Dpb11 | DNA replication initiation protein. Loads DNA polymerase ε onto pre-replication complexes at origins. |
| Fen1 | 5' flap endonuclease involved in processing Okazaki fragments. |
| Geminin | Protein found in metazoans and absent from yeasts. Binds to and inactivates Cdt1, thereby regulating pre-replicative/initiation complex formation. Also suggested to promote pre-RC formation by binding and thus preventing Cdt1 degradation |
| GINS | Tetrameric complex composed of Sld5, Psf1, Psf2, Psf3. Associates with pre-replicative complex around the time of initiation and moves with replication forks during elongation step. Required for elongation stage of DNA replication and maybe part of the Mcm helicase complex. |
| Minichromosome maintenance proteins (Mcm) | Six different proteins of the AAA+ ATPase family that form a hexamer in solution. This hexamer is recruited and loaded by ORC, Cdc6 and Cdt1 and forms a double hexamer that is topologically linked around DNA to form a salt-resistant pre-replicative complex. On replication initiation, Mcm2-7 moves away from ORC with replication fork. |
| Mcm10 | Required for initiation and elongation stages of DNA replication. Implicated in chromatin binding of Cdc45 and DNA polymerase α. Also required for stability of DNA polymerase α catalytic subunit in the budding yeast S. cerevisiae. |
| Mrc1 | Couple leading-strand synthesis with the CMG complex helicase activity. Metazoan homolog is known as Claspin. |
| Origin recognition complex (ORC) | Heterohexameric complex composed of Orc1–Orc6 proteins. Binds to DNA and assembles Mcm2-7 complex onto chromatin together with Cdc6 and Cdt1. |
| Proliferating cell nuclear antigen (PCNA) | Trimeric protein with ring shaped structure, encloses DNA preventing dissociation of DNA polymerase. Acts as a sliding clamp for polymerases δ and ε, thereby improving processivity of replicative polymerases. |
| Replication factor C (RFC) | Loads PCNA on primed templates and is involved in the switch between DNA polymerase a and the replicative polymerases δ and ε. |
| Replication fork barriers (RFBs) | Bound by RFB proteins in various locations throughout the genome. Are able to terminate or pause replication forks, stopping progression of the replisome. |
| Replication protein A (RPA) | Heterotrimeric single-stranded binding protein. Stabilizes single-stranded DNA at replication fork. |
| RNase H | Ribonuclease which digests RNA hybridized to DNA. Involved in Okazaki fragment processing. |
| Sld2 | Functions in initiation of replication. Key substrate of CDK, phosphorylation promotes interaction with Dpb11. Required for initiation of replication. |
| Sld3 | Functions in initiation of replication. Key substrate of CDK, phosphorylation promotes interaction with Dpb11. Required for initiation of replication. |
| Telomerase | A ribonucleoprotein that adds DNA sequence "TTAGGG" repeats to the 3' end of DNA strands in telomeres. |
| Topoisomerases | Regulate the overwinding or underwinding of DNA |
References
- Leman AR, Noguchi E (March 2013). "The replication fork: understanding the eukaryotic replication machinery and the challenges to genome duplication". Genes. 4 (1): 1–32. doi:10.3390/genes4010001. PMC 3627427. PMID 23599899.
- Blow JJ, Dutta A (June 2005). "Preventing re-replication of chromosomal DNA". Nature Reviews Molecular Cell Biology. 6 (6): 476–86. doi:10.1038/nrm1663. PMC 2688777. PMID 15928711.
- Fisher PA, Wang TS, Korn D (July 1979). "Enzymological characterization of DNA polymerase alpha. Basic catalytic properties processivity, and gap utilization of the homogeneous enzyme from human KB cells". The Journal of Biological Chemistry. 254 (13): 6128–37. doi:10.1016/S0021-9258(18)50528-7. PMID 447699.
- Boos D, Ferreira P (March 2019). "Origin Firing Regulations to Control Genome Replication Timing". Genes. 10 (3): 199. doi:10.3390/genes10030199. PMC 6470937. PMID 30845782.
- Foss EJ, Lichauco C, Gatbonton-Schwager T, Lofts B, Lao U, Bedalov A (2023-07-03). "Identification of 1600 replication origins in S. cerevisiae". eLife. 12. doi:10.7554/eLife.88087.
- Araki H (2011). "Initiation of chromosomal DNA replication in eukaryotic cells; contribution of yeast genetics to the elucidation". Genes & Genetic Systems. 86 (3): 141–9. doi:10.1266/ggs.86.141. PMID 21952204.
- Maiorano D, Moreau J, Méchali M (April 2000). "XCDT1 is required for the assembly of pre-replicative complexes in Xenopus laevis". Nature. 404 (6778): 622–5. Bibcode:2000Natur.404..622M. doi:10.1038/35007104. PMID 10766247. S2CID 4416138.
- Bell SP, Dutta A (2002). "DNA replication in eukaryotic cells". Annual Review of Biochemistry. 71: 333–74. doi:10.1146/annurev.biochem.71.110601.135425. PMID 12045100.
- Tye BK (1999). "MCM proteins in DNA replication". Annual Review of Biochemistry. 68 (1): 649–86. doi:10.1146/annurev.biochem.68.1.649. PMID 10872463.
- Ticau S, Friedman LJ, Ivica NA, Gelles J, Bell SP (April 2015). "Single-molecule studies of origin licensing reveal mechanisms ensuring bidirectional helicase loading". Cell. 161 (3): 513–525. doi:10.1016/j.cell.2015.03.012. PMC 4445235. PMID 25892223.
- Zhai Y, Li N, Jiang H, Huang X, Gao N, Tye BK (July 2017). "Unique Roles of the Non-identical MCM Subunits in DNA Replication Licensing". Molecular Cell. 67 (2): 168–179. doi:10.1016/j.molcel.2017.06.016. PMID 28732205.
- Coster G, Diffley JF (July 2017). "Bidirectional eukaryotic DNA replication is established by quasi-symmetrical helicase loading". Science. 357 (6348): 314–318. Bibcode:2017Sci...357..314C. doi:10.1126/science.aan0063. PMC 5608077. PMID 28729513.
- Zhai Y, Cheng E, Wu H, Li N, Yung PY, Gao N, Tye BK (March 2017). "Open-ringed structure of the Cdt1-Mcm2-7 complex as a precursor of the MCM double hexamer". Nature Structural & Molecular Biology. 24 (3): 300–308. doi:10.1038/nsmb.3374. PMID 28191894. S2CID 3929807.
- Bell SP (March 2002). "The origin recognition complex: from simple origins to complex functions". Genes & Development. 16 (6): 659–72. doi:10.1101/gad.969602. PMID 11914271.
- Bell SP, Stillman B (May 1992). "ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex". Nature. 357 (6374): 128–134. Bibcode:1992Natur.357..128B. doi:10.1038/357128a0. PMID 1579162. S2CID 4346767.
- Li N, Lam WH, Zhai Y, Cheng J, Cheng E, Zhao Y, et al. (July 2018). "Structure of the origin recognition complex bound to DNA replication origin". Nature. 559 (7713): 217–222. Bibcode:2018Natur.559..217L. doi:10.1038/s41586-018-0293-x. PMID 29973722. S2CID 49577101.
- Miotto B, Ji Z, Struhl K (August 2016). "Selectivity of ORC binding sites and the relation to replication timing, fragile sites, and deletions in cancers". Proceedings of the National Academy of Sciences of the United States of America. 113 (33): E4810-9. Bibcode:2016PNAS..113E4810M. doi:10.1073/pnas.1609060113. PMC 4995967. PMID 27436900.
- Lee CS, Cheung MF, Li J, Zhao Y, Lam WH, Ho V, et al. (January 2021). "Humanizing the yeast origin recognition complex". Nature Communications. 12 (1): 33. Bibcode:2021NatCo..12...33L. doi:10.1038/s41467-020-20277-y. PMC 7782691. PMID 33397927.
- Bleichert F, Leitner A, Aebersold R, Botchan MR, Berger JM (June 2018). "Conformational control and DNA-binding mechanism of the metazoan origin recognition complex". Proceedings of the National Academy of Sciences of the United States of America. 115 (26): E5906–E5915. Bibcode:2018PNAS..115E5906B. doi:10.1073/pnas.1806315115. PMC 6042147. PMID 29899147.
- Chesnokov IN (2007). "Multiple functions of the origin recognition complex". International Review of Cytology. 256: 69–109. doi:10.1016/S0074-7696(07)56003-1. ISBN 9780123737007. PMID 17241905.
- Matsuda K, Makise M, Sueyasu Y, Takehara M, Asano T, Mizushima T (December 2007). "Yeast two-hybrid analysis of the origin recognition complex of Saccharomyces cerevisiae: interaction between subunits and identification of binding proteins". FEMS Yeast Research. 7 (8): 1263–9. doi:10.1111/j.1567-1364.2007.00298.x. PMID 17825065.
- Kreitz S, Ritzi M, Baack M, Knippers R (March 2001). "The human origin recognition complex protein 1 dissociates from chromatin during S phase in HeLa cells". The Journal of Biological Chemistry. 276 (9): 6337–42. doi:10.1074/jbc.M009473200. PMID 11102449.
- Speck C, Chen Z, Li H, Stillman B (November 2005). "ATPase-dependent cooperative binding of ORC and Cdc6 to origin DNA". Nature Structural & Molecular Biology. 12 (11): 965–71. doi:10.1038/nsmb1002. PMC 2952294. PMID 16228006.
- Coleman TR, Carpenter PB, Dunphy WG (October 1996). "The Xenopus Cdc6 protein is essential for the initiation of a single round of DNA replication in cell-free extracts". Cell. 87 (1): 53–63. doi:10.1016/S0092-8674(00)81322-7. PMID 8858148. S2CID 16897247.
- Rialland M, Sola F, Santocanale C (April 2002). "Essential role of human CDT1 in DNA replication and chromatin licensing". Journal of Cell Science. 115 (Pt 7): 1435–40. doi:10.1242/jcs.115.7.1435. PMID 11896191.
- Tanaka S, Diffley JF (March 2002). "Interdependent nuclear accumulation of budding yeast Cdt1 and Mcm2-7 during G1 phase". Nature Cell Biology. 4 (3): 198–207. doi:10.1038/ncb757. PMID 11836525. S2CID 45861829.
- Frigola J, He J, Kinkelin K, Pye VE, Renault L, Douglas ME, Remus D, Cherepanov P, Costa A, Diffley JF (June 2017). "Cdt1 stabilizes an open MCM ring for helicase loading". Nature Communications. 8: 15720. Bibcode:2017NatCo...815720F. doi:10.1038/ncomms15720. PMC 5490006. PMID 28643783.
- Ticau S, Friedman LJ, Champasa K, Corrêa IR, Gelles J, Bell SP (March 2017). "Mechanism and timing of Mcm2-7 ring closure during DNA replication origin licensing". Nature Structural & Molecular Biology. 24 (3): 309–315. doi:10.1038/nsmb.3375. PMC 5336523. PMID 28191892.
- Nishitani H, Lygerou Z, Nishimoto T, Nurse P (April 2000). "The Cdt1 protein is required to license DNA for replication in fission yeast". Nature. 404 (6778): 625–8. Bibcode:2000Natur.404..625N. doi:10.1038/35007110. PMID 10766248. S2CID 205005540.
- Yuan Z, Riera A, Bai L, Sun J, Nandi S, Spanos C, Chen ZA, Barbon M, Rappsilber J, Stillman B, Speck C, Li H (March 2017). "Structural basis of Mcm2-7 replicative helicase loading by ORC-Cdc6 and Cdt1". Nature Structural & Molecular Biology. 24 (3): 316–324. doi:10.1038/nsmb.3372. PMC 5503505. PMID 28191893.
- Wohlschlegel JA, Dwyer BT, Dhar SK, Cvetic C, Walter JC, Dutta A (December 2000). "Inhibition of eukaryotic DNA replication by geminin binding to Cdt1". Science. 290 (5500): 2309–12. Bibcode:2000Sci...290.2309W. doi:10.1126/science.290.5500.2309. PMID 11125146.
- Maine GT, Sinha P, Tye BK (March 1984). "Mutants of S. cerevisiae defective in the maintenance of minichromosomes". Genetics. 106 (3): 365–85. doi:10.1093/genetics/106.3.365. PMC 1224244. PMID 6323245.
- Hua XH, Newport J (January 1998). "Identification of a preinitiation step in DNA replication that is independent of origin recognition complex and cdc6, but dependent on cdk2". The Journal of Cell Biology. 140 (2): 271–81. doi:10.1083/jcb.140.2.271. PMC 2132576. PMID 9442103.
- Rowles A, Tada S, Blow JJ (June 1999). "Changes in association of the Xenopus origin recognition complex with chromatin on licensing of replication origins". Journal of Cell Science. 112 (12): 2011–8. doi:10.1242/jcs.112.12.2011. PMC 3605702. PMID 10341218.
- Li N, Zhai Y, Zhang Y, Li W, Yang M, Lei J, Tye BK, Gao N (August 2015). "Structure of the eukaryotic MCM complex at 3.8 Å". Nature. 524 (7564): 186–91. Bibcode:2015Natur.524..186L. doi:10.1038/nature14685. PMID 26222030. S2CID 4468690.
- Noguchi Y, Yuan Z, Bai L, Schneider S, Zhao G, Stillman B, Speck C, Li H (November 2017). "Cryo-EM structure of Mcm2-7 double hexamer on DNA suggests a lagging-strand DNA extrusion model". Proceedings of the National Academy of Sciences of the United States of America. 114 (45): E9529–E9538. Bibcode:2017PNAS..114E9529N. doi:10.1073/pnas.1712537114. PMC 5692578. PMID 29078375.
- Dutta A, Bell SP (1997). "Initiation of DNA replication in eukaryotic cells". Annual Review of Cell and Developmental Biology. 13: 293–332. doi:10.1146/annurev.cellbio.13.1.293. PMID 9442876.
- Labib K, Tercero JA, Diffley JF (June 2000). "Uninterrupted MCM2-7 function required for DNA replication fork progression". Science. 288 (5471): 1643–7. Bibcode:2000Sci...288.1643L. doi:10.1126/science.288.5471.1643. PMID 10834843.
- Schwacha A, Bell SP (November 2001). "Interactions between two catalytically distinct MCM subgroups are essential for coordinated ATP hydrolysis and DNA replication". Molecular Cell. 8 (5): 1093–104. doi:10.1016/S1097-2765(01)00389-6. PMID 11741544.
- Bochman ML, Bell SP, Schwacha A (October 2008). "Subunit organization of Mcm2-7 and the unequal role of active sites in ATP hydrolysis and viability". Molecular and Cellular Biology. 28 (19): 5865–5873. doi:10.1128/MCB.00161-08. PMC 2547011. PMID 18662997.
- Hingorani MM, Washington MT, Moore KC, Patel SS (May 1997). "The dTTPase mechanism of T7 DNA helicase resembles the binding change mechanism of the F1-ATPase". Proceedings of the National Academy of Sciences of the United States of America. 94 (10): 5012–7. Bibcode:1997PNAS...94.5012H. doi:10.1073/pnas.94.10.5012. PMC 24622. PMID 9144181.
- Yan H, Merchant AM, Tye BK (November 1993). "Cell cycle-regulated nuclear localization of MCM2 and MCM3, which are required for the initiation of DNA synthesis at chromosomal replication origins in yeast". Genes & Development. 7 (11): 2149–60. doi:10.1101/gad.7.11.2149. PMID 8224843.
- Young MR, Suzuki K, Yan H, Gibson S, Tye BK (October 1997). "Nuclear accumulation of Saccharomyces cerevisiae Mcm3 is dependent on its nuclear localization sequence". Genes to Cells. 2 (10): 631–43. doi:10.1046/j.1365-2443.1997.1510349.x. PMID 9427284.
- Labib K, Diffley JF, Kearsey SE (November 1999). "G1-phase and B-type cyclins exclude the DNA-replication factor Mcm4 from the nucleus". Nature Cell Biology. 1 (7): 415–22. doi:10.1038/15649. PMID 10559985. S2CID 23407351.
- Lei M, Tye BK (April 2001). "Initiating DNA synthesis: from recruiting to activating the MCM complex". Journal of Cell Science. 114 (Pt 8): 1447–54. doi:10.1242/jcs.114.8.1447. PMID 11282021.
- Abid Ali F, Douglas ME, Locke J, Pye VE, Nans A, Diffley JF, Costa A (December 2017). "Cryo-EM structure of a licensed DNA replication origin". Nature Communications. 8 (1): 2241. Bibcode:2017NatCo...8.2241A. doi:10.1038/s41467-017-02389-0. PMC 5740162. PMID 29269875.
- Li J, Dong J, Wang W, Yu D, Fan X, Hui YC, et al. (January 2023). "The human pre-replication complex is an open complex" (PDF). Cell. 186 (1): 98–111.e21. doi:10.1016/j.cell.2022.12.008. PMID 36608662. S2CID 255442139.
- Greiwe JF, Miller TC, Locke J, Martino F, Howell S, Schreiber A, et al. (January 2022). "Structural mechanism for the selective phosphorylation of DNA-loaded MCM double hexamers by the Dbf4-dependent kinase". Nature Structural & Molecular Biology. 29 (1): 10–20. doi:10.1038/s41594-021-00698-z. PMC 8770131. PMID 34963704.
- Saleh A, Noguchi Y, Aramayo R, Ivanova ME, Stevens KM, Montoya A, et al. (May 2022). "The structural basis of Cdc7-Dbf4 kinase dependent targeting and phosphorylation of the MCM2-7 double hexamer". Nature Communications. 13 (1): 2915. Bibcode:2022NatCo..13.2915S. doi:10.1038/s41467-022-30576-1. PMC 9133112. PMID 35614055.
- Cheng J, Li N, Huo Y, Dang S, Tye BK, Gao N, Zhai Y (March 2022). "Structural Insight into the MCM double hexamer activation by Dbf4-Cdc7 kinase". Nature Communications. 13 (1): 1396. Bibcode:2022NatCo..13.1396C. doi:10.1038/s41467-022-29070-5. PMC 8927117. PMID 35296675.
- Sheu YJ, Stillman B (January 2010). "The Dbf4-Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4". Nature. 463 (7277): 113–117. Bibcode:2010Natur.463..113S. doi:10.1038/nature08647. PMC 2805463. PMID 20054399.
- Tercero JA, Labib K, Diffley JF (May 2000). "DNA synthesis at individual replication forks requires the essential initiation factor Cdc45p". The EMBO Journal. 19 (9): 2082–93. doi:10.1093/emboj/19.9.2082. PMC 305696. PMID 10790374.
- Hennessy KM, Lee A, Chen E, Botstein D (June 1991). "A group of interacting yeast DNA replication genes". Genes & Development. 5 (6): 958–69. doi:10.1101/gad.5.6.958. PMID 2044962.
- Aparicio OM, Weinstein DM, Bell SP (October 1997). "Components and dynamics of DNA replication complexes in S. cerevisiae: redistribution of MCM proteins and Cdc45p during S phase". Cell. 91 (1): 59–69. doi:10.1016/S0092-8674(01)80009-X. PMID 9335335. S2CID 10353164.
- Mimura S, Masuda T, Matsui T, Takisawa H (June 2000). "Central role for cdc45 in establishing an initiation complex of DNA replication in Xenopus egg extracts". Genes to Cells. 5 (6): 439–52. doi:10.1046/j.1365-2443.2000.00340.x. PMID 10886370.
- Aparicio OM, Stout AM, Bell SP (August 1999). "Differential assembly of Cdc45p and DNA polymerases at early and late origins of DNA replication". Proceedings of the National Academy of Sciences of the United States of America. 96 (16): 9130–5. Bibcode:1999PNAS...96.9130A. doi:10.1073/pnas.96.16.9130. PMC 17744. PMID 10430907.
- Zou L, Stillman B (May 2000). "Assembly of a complex containing Cdc45p, replication protein A, and Mcm2p at replication origins controlled by S-phase cyclin-dependent kinases and Cdc7p-Dbf4p kinase". Molecular and Cellular Biology. 20 (9): 3086–96. doi:10.1128/mcb.20.9.3086-3096.2000. PMC 85601. PMID 10757793.
- Walter J, Newport J (April 2000). "Initiation of eukaryotic DNA replication: origin unwinding and sequential chromatin association of Cdc45, RPA, and DNA polymerase alpha". Molecular Cell. 5 (4): 617–27. doi:10.1016/S1097-2765(00)80241-5. PMID 10882098.
- Gambus A, Jones RC, Sanchez-Diaz A, Kanemaki M, van Deursen F, Edmondson RD, Labib K (April 2006). "GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks". Nature Cell Biology. 8 (4): 358–66. doi:10.1038/ncb1382. PMID 16531994. S2CID 21543095.
- Kanemaki M, Sanchez-Diaz A, Gambus A, Labib K (June 2003). "Functional proteomic identification of DNA replication proteins by induced proteolysis in vivo". Nature. 423 (6941): 720–4. Bibcode:2003Natur.423..720K. doi:10.1038/nature01692. PMID 12768207. S2CID 4345091.
- Makarova KS, Wolf YI, Mekhedov SL, Mirkin BG, Koonin EV (2005). "Ancestral paralogs and pseudoparalogs and their role in the emergence of the eukaryotic cell". Nucleic Acids Research. 33 (14): 4626–38. doi:10.1093/nar/gki775. PMC 1187821. PMID 16106042.
- Moyer SE, Lewis PW, Botchan MR (July 2006). "Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase". Proceedings of the National Academy of Sciences of the United States of America. 103 (27): 10236–10241. Bibcode:2006PNAS..10310236M. doi:10.1073/pnas.0602400103. PMC 1482467. PMID 16798881.
- Bochman ML, Schwacha A (July 2008). "The Mcm2-7 complex has in vitro helicase activity". Molecular Cell. 31 (2): 287–93. doi:10.1016/j.molcel.2008.05.020. PMID 18657510.
- Costa A, Ilves I, Tamberg N, Petojevic T, Nogales E, Botchan MR, Berger JM (April 2011). "The structural basis for MCM2-7 helicase activation by GINS and Cdc45". Nature Structural & Molecular Biology. 18 (4): 471–7. doi:10.1038/nsmb.2004. PMC 4184033. PMID 21378962.
- Yuan Z, Bai L, Sun J, Georgescu R, Liu J, O'Donnell ME, Li H (March 2016). "Structure of the eukaryotic replicative CMG helicase suggests a pumpjack motion for translocation". Nature Structural & Molecular Biology. 23 (3): 217–24. doi:10.1038/nsmb.3170. PMC 4812828. PMID 26854665.
- Homesley L, Lei M, Kawasaki Y, Sawyer S, Christensen T, Tye BK (April 2000). "Mcm10 and the MCM2-7 complex interact to initiate DNA synthesis and to release replication factors from origins". Genes & Development. 14 (8): 913–26. doi:10.1101/gad.14.8.913. PMC 316538. PMID 10783164.
- Christensen TW, Tye BK (June 2003). "Drosophila MCM10 interacts with members of the prereplication complex and is required for proper chromosome condensation". Molecular Biology of the Cell. 14 (6): 2206–15. doi:10.1091/mbc.e02-11-0706. PMC 194871. PMID 12808023.
- Lee C, Liachko I, Bouten R, Kelman Z, Tye BK (January 2010). "Alternative mechanisms for coordinating polymerase alpha and MCM helicase". Molecular and Cellular Biology. 30 (2): 423–35. doi:10.1128/MCB.01240-09. PMC 2798462. PMID 19917723.
- van Deursen F, Sengupta S, De Piccoli G, Sanchez-Diaz A, Labib K (May 2012). "Mcm10 associates with the loaded DNA helicase at replication origins and defines a novel step in its activation". The EMBO Journal. 31 (9): 2195–206. doi:10.1038/emboj.2012.69. PMC 3343467. PMID 22433841.
- Masai H, Sato N, Takeda T, Arai K (December 1999). "CDC7 kinase complex as a molecular switch for DNA replication". Frontiers in Bioscience. 4 (1–3): D834-40. doi:10.2741/masai. PMID 10577390.
- Mimura S, Takisawa H (October 1998). "Xenopus Cdc45-dependent loading of DNA polymerase alpha onto chromatin under the control of S-phase Cdk". The EMBO Journal. 17 (19): 5699–707. doi:10.1093/emboj/17.19.5699. PMC 1170898. PMID 9755170.
- Zou L, Stillman B (April 1998). "Formation of a preinitiation complex by S-phase cyclin CDK-dependent loading of Cdc45p onto chromatin". Science. 280 (5363): 593–6. Bibcode:1998Sci...280..593Z. doi:10.1126/science.280.5363.593. PMID 9554851.
- Jiang W, Wells NJ, Hunter T (May 1999). "Multistep regulation of DNA replication by Cdk phosphorylation of HsCdc6". Proceedings of the National Academy of Sciences of the United States of America. 96 (11): 6193–8. Bibcode:1999PNAS...96.6193J. doi:10.1073/pnas.96.11.6193. PMC 26858. PMID 10339564.
- Jiang W, McDonald D, Hope TJ, Hunter T (October 1999). "Mammalian Cdc7-Dbf4 protein kinase complex is essential for initiation of DNA replication". The EMBO Journal. 18 (20): 5703–5713. doi:10.1093/emboj/18.20.5703. PMC 1171637. PMID 10523313.
- Kumagai H, Sato N, Yamada M, Mahony D, Seghezzi W, Lees E, et al. (July 1999). "A novel growth- and cell cycle-regulated protein, ASK, activates human Cdc7-related kinase and is essential for G1/S transition in mammalian cells". Molecular and Cellular Biology. 19 (7): 5083–5095. doi:10.1128/MCB.19.7.5083. PMC 84351. PMID 10373557.
- Francis LI, Randell JC, Takara TJ, Uchima L, Bell SP (March 2009). "Incorporation into the prereplicative complex activates the Mcm2-7 helicase for Cdc7-Dbf4 phosphorylation". Genes & Development. 23 (5): 643–654. doi:10.1101/gad.1759609. PMC 2658526. PMID 19270162.
- Sun J, Fernandez-Cid A, Riera A, Tognetti S, Yuan Z, Stillman B, et al. (October 2014). "Structural and mechanistic insights into Mcm2-7 double-hexamer assembly and function". Genes & Development. 28 (20): 2291–2303. doi:10.1101/gad.242313.114. PMC 4201289. PMID 25319829.
- Lei M, Kawasaki Y, Young MR, Kihara M, Sugino A, Tye BK (December 1997). "Mcm2 is a target of regulation by Cdc7-Dbf4 during the initiation of DNA synthesis". Genes & Development. 11 (24): 3365–3374. doi:10.1101/gad.11.24.3365. PMC 316824. PMID 9407029.
- Kamimura Y, Tak YS, Sugino A, Araki H (April 2001). "Sld3, which interacts with Cdc45 (Sld4), functions for chromosomal DNA replication in Saccharomyces cerevisiae". The EMBO Journal. 20 (8): 2097–107. doi:10.1093/emboj/20.8.2097. PMC 125422. PMID 11296242.
- Kanemaki M, Labib K (April 2006). "Distinct roles for Sld3 and GINS during establishment and progression of eukaryotic DNA replication forks". The EMBO Journal. 25 (8): 1753–63. doi:10.1038/sj.emboj.7601063. PMC 1440835. PMID 16601689.
- Masumoto H, Sugino A, Araki H (April 2000). "Dpb11 controls the association between DNA polymerases alpha and epsilon and the autonomously replicating sequence region of budding yeast". Molecular and Cellular Biology. 20 (8): 2809–17. doi:10.1128/mcb.20.8.2809-2817.2000. PMC 85497. PMID 10733584.
- Araki H, Leem SH, Phongdara A, Sugino A (December 1995). "Dpb11, which interacts with DNA polymerase II(epsilon) in Saccharomyces cerevisiae, has a dual role in S-phase progression and at a cell cycle checkpoint". Proceedings of the National Academy of Sciences of the United States of America. 92 (25): 11791–5. doi:10.1073/pnas.92.25.11791. PMC 40488. PMID 8524850.
- Glover JN, Williams RS, Lee MS (November 2004). "Interactions between BRCT repeats and phosphoproteins: tangled up in two". Trends in Biochemical Sciences. 29 (11): 579–85. doi:10.1016/j.tibs.2004.09.010. PMID 15501676.
- Masumoto H, Muramatsu S, Kamimura Y, Araki H (February 2002). "S-Cdk-dependent phosphorylation of Sld2 essential for chromosomal DNA replication in budding yeast". Nature. 415 (6872): 651–5. Bibcode:2002Natur.415..651M. doi:10.1038/nature713. PMID 11807498. S2CID 4300863.
- Takayama Y, Kamimura Y, Okawa M, Muramatsu S, Sugino A, Araki H (May 2003). "GINS, a novel multiprotein complex required for chromosomal DNA replication in budding yeast". Genes & Development. 17 (9): 1153–65. doi:10.1101/gad.1065903. PMC 196052. PMID 12730134.
- Labib K, Gambus A (June 2007). "A key role for the GINS complex at DNA replication forks". Trends in Cell Biology. 17 (6): 271–8. doi:10.1016/j.tcb.2007.04.002. PMID 17467990.
- Muramatsu S, Hirai K, Tak YS, Kamimura Y, Araki H (March 2010). "CDK-dependent complex formation between replication proteins Dpb11, Sld2, Pol (epsilon), and GINS in budding yeast". Genes & Development. 24 (6): 602–12. doi:10.1101/gad.1883410. PMC 2841337. PMID 20231317.
- Lehman IR, Bessman MJ, Simms ES, Kornberg A (July 1958). "Enzymatic synthesis of deoxyribonucleic acid. I. Preparation of substrates and partial purification of an enzyme from Escherichia coli". The Journal of Biological Chemistry. 233 (1): 163–70. doi:10.1016/S0021-9258(19)68048-8. PMID 13563462.
- Alani E, Thresher R, Griffith JD, Kolodner RD (September 1992). "Characterization of DNA-binding and strand-exchange stimulation properties of y-RPA, a yeast single-strand-DNA-binding protein". Journal of Molecular Biology. 227 (1): 54–71. doi:10.1016/0022-2836(92)90681-9. PMID 1522601.
- Goulian M, Richards SH, Heard CJ, Bigsby BM (October 1990). "Discontinuous DNA synthesis by purified mammalian proteins". The Journal of Biological Chemistry. 265 (30): 18461–71. doi:10.1016/S0021-9258(17)44775-2. PMID 2170412.
- McCulloch SD, Kunkel TA (January 2008). "The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases". Cell Research. 18 (1): 148–61. doi:10.1038/cr.2008.4. PMC 3639319. PMID 18166979.
- Pursell ZF, Isoz I, Lundström EB, Johansson E, Kunkel TA (July 2007). "Yeast DNA polymerase epsilon participates in leading-strand DNA replication". Science. 317 (5834): 127–30. Bibcode:2007Sci...317..127P. doi:10.1126/science.1144067. PMC 2233713. PMID 17615360.
- Watson J, Baker T, Bell S, Gann A, Levine M, Losick R, Molecular Biology of the Gene 6th Edition, Pearson Education Inc, 2008. ISBN 080539592X
- Waga S, Bauer G, Stillman B (April 1994). "Reconstitution of complete SV40 DNA replication with purified replication factors". The Journal of Biological Chemistry. 269 (14): 10923–34. doi:10.1016/S0021-9258(17)34146-7. PMID 8144677.
- Garg P, Stith CM, Sabouri N, Johansson E, Burgers PM (November 2004). "Idling by DNA polymerase delta maintains a ligatable nick during lagging-strand DNA replication". Genes & Development. 18 (22): 2764–73. doi:10.1101/gad.1252304. PMC 528896. PMID 15520275.
- Burgers PM (February 2009). "Polymerase dynamics at the eukaryotic DNA replication fork". The Journal of Biological Chemistry. 284 (7): 4041–5. doi:10.1074/jbc.R800062200. PMC 2640984. PMID 18835809.
- Jin YH, Ayyagari R, Resnick MA, Gordenin DA, Burgers PM (January 2003). "Okazaki fragment maturation in yeast. II. Cooperation between the polymerase and 3'-5'-exonuclease activities of Pol delta in the creation of a ligatable nick". The Journal of Biological Chemistry. 278 (3): 1626–33. doi:10.1074/jbc.M209803200. PMID 12424237.
- Sogo JM, Lopes M, Foiani M (July 2002). "Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects". Science. 297 (5581): 599–602. Bibcode:2002Sci...297..599S. doi:10.1126/science.1074023. PMID 12142537. S2CID 33502697.
- Morrison A, Araki H, Clark AB, Hamatake RK, Sugino A (September 1990). "A third essential DNA polymerase in S. cerevisiae". Cell. 62 (6): 1143–51. doi:10.1016/0092-8674(90)90391-Q. PMID 2169349. S2CID 29672985.
- Waga S, Stillman B (May 1994). "Anatomy of a DNA replication fork revealed by reconstitution of SV40 DNA replication in vitro". Nature. 369 (6477): 207–12. Bibcode:1994Natur.369..207W. doi:10.1038/369207a0. PMID 7910375. S2CID 4351628.
- Tsurimoto T, Stillman B (January 1991). "Replication factors required for SV40 DNA replication in vitro. II. Switching of DNA polymerase alpha and delta during initiation of leading and lagging strand synthesis". The Journal of Biological Chemistry. 266 (3): 1961–8. doi:10.1016/S0021-9258(18)52386-3. PMID 1671046.
- Barry ER, Bell SD (December 2006). "DNA replication in the archaea". Microbiology and Molecular Biology Reviews. 70 (4): 876–87. doi:10.1128/MMBR.00029-06. PMC 1698513. PMID 17158702.
- Berg JM, Tymoczko JL, Stryer L (2003). Biochemie. Heidelberg/Berlin: Springer.
- Johnson RE, Klassen R, Prakash L, Prakash S (July 2015). "A Major Role of DNA Polymerase δ in Replication of Both the Leading and Lagging DNA Strands". Molecular Cell. 59 (2): 163–175. doi:10.1016/j.molcel.2015.05.038. PMC 4517859. PMID 26145172.
- Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA (May 2005). "Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint". Genes & Development. 19 (9): 1040–52. doi:10.1101/gad.1301205. PMC 1091739. PMID 15833913.
- Remus D, Beuron F, Tolun G, Griffith JD, Morris EP, Diffley JF (November 2009). "Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing". Cell. 139 (4): 719–30. doi:10.1016/j.cell.2009.10.015. PMC 2804858. PMID 19896182.
- Evrin C, Clarke P, Zech J, Lurz R, Sun J, Uhle S, Li H, Stillman B, Speck C (December 2009). "A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication". Proceedings of the National Academy of Sciences of the United States of America. 106 (48): 20240–5. Bibcode:2009PNAS..10620240E. doi:10.1073/pnas.0911500106. PMC 2787165. PMID 19910535.
- Labib K (June 2010). "How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells?". Genes & Development. 24 (12): 1208–19. doi:10.1101/gad.1933010. PMC 2885657. PMID 20551170.
- Pacek M, Walter JC (September 2004). "A requirement for MCM7 and Cdc45 in chromosome unwinding during eukaryotic DNA replication". The EMBO Journal. 23 (18): 3667–76. doi:10.1038/sj.emboj.7600369. PMC 517609. PMID 15329670.
- Yao NY, O'Donnell M (June 2010). "SnapShot: The replisome". Cell. 141 (6): 1088–1088.e1. doi:10.1016/j.cell.2010.05.042. PMC 4007198. PMID 20550941.
- Bermudez VP, Farina A, Tappin I, Hurwitz J (March 2010). "Influence of the human cohesion establishment factor Ctf4/AND-1 on DNA replication". The Journal of Biological Chemistry. 285 (13): 9493–505. doi:10.1074/jbc.M109.093609. PMC 2843200. PMID 20089864.
- Zhu W, Ukomadu C, Jha S, Senga T, Dhar SK, Wohlschlegel JA, Nutt LK, Kornbluth S, Dutta A (September 2007). "Mcm10 and And-1/CTF4 recruit DNA polymerase alpha to chromatin for initiation of DNA replication". Genes & Development. 21 (18): 2288–99. doi:10.1101/gad.1585607. PMC 1973143. PMID 17761813.
- Lou H, Komata M, Katou Y, Guan Z, Reis CC, Budd M, Shirahige K, Campbell JL (October 2008). "Mrc1 and DNA polymerase epsilon function together in linking DNA replication and the S phase checkpoint". Molecular Cell. 32 (1): 106–17. doi:10.1016/j.molcel.2008.08.020. PMC 2699584. PMID 18851837.
- Petermann E, Helleday T, Caldecott KW (June 2008). "Claspin promotes normal replication fork rates in human cells". Molecular Biology of the Cell. 19 (6): 2373–8. doi:10.1091/mbc.E07-10-1035. PMC 2397295. PMID 18353973.
- Bravo R, Frank R, Blundell PA, Macdonald-Bravo H (1987). "Cyclin/PCNA is the auxiliary protein of DNA polymerase-delta". Nature. 326 (6112): 515–7. Bibcode:1987Natur.326..515B. doi:10.1038/326515a0. PMID 2882423. S2CID 4344147.
- Prelich G, Tan CK, Kostura M, Mathews MB, So AG, Downey KM, Stillman B (1987). "Functional identity of proliferating cell nuclear antigen and a DNA polymerase-delta auxiliary protein". Nature. 326 (6112): 517–20. Bibcode:1987Natur.326..517P. doi:10.1038/326517a0. PMID 2882424. S2CID 4345171.
- Moldovan GL, Pfander B, Jentsch S (May 2007). "PCNA, the maestro of the replication fork". Cell. 129 (4): 665–79. doi:10.1016/j.cell.2007.05.003. PMID 17512402. S2CID 3547069.
- Cai J, Yao N, Gibbs E, Finkelstein J, Phillips B, O'Donnell M, Hurwitz J (September 1998). "ATP hydrolysis catalyzed by human replication factor C requires participation of multiple subunits". Proceedings of the National Academy of Sciences of the United States of America. 95 (20): 11607–12. Bibcode:1998PNAS...9511607C. doi:10.1073/pnas.95.20.11607. PMC 21688. PMID 9751713.
- Tsurimoto T, Stillman B (January 1991). "Replication factors required for SV40 DNA replication in vitro. I. DNA structure-specific recognition of a primer-template junction by eukaryotic DNA polymerases and their accessory proteins". The Journal of Biological Chemistry. 266 (3): 1950–60. doi:10.1016/S0021-9258(18)52385-1. PMID 1671045.
- Podust LM, Podust VN, Sogo JM, Hübscher U (June 1995). "Mammalian DNA polymerase auxiliary proteins: analysis of replication factor C-catalyzed proliferating cell nuclear antigen loading onto circular double-stranded DNA". Molecular and Cellular Biology. 15 (6): 3072–81. doi:10.1128/MCB.15.6.3072. PMC 230538. PMID 7760803.
- Zhang G, Gibbs E, Kelman Z, O'Donnell M, Hurwitz J (March 1999). "Studies on the interactions between human replication factor C and human proliferating cell nuclear antigen". Proceedings of the National Academy of Sciences of the United States of America. 96 (5): 1869–74. Bibcode:1999PNAS...96.1869Z. doi:10.1073/pnas.96.5.1869. PMC 26703. PMID 10051561.
- Yao NY, Johnson A, Bowman GD, Kuriyan J, O'Donnell M (June 2006). "Mechanism of proliferating cell nuclear antigen clamp opening by replication factor C". The Journal of Biological Chemistry. 281 (25): 17528–39. doi:10.1074/jbc.M601273200. PMID 16608854.
- Sabatinos, SA (2010). "Recovering a Stalled Replication Fork". Scitable Nature Education. 3 (9): 31.
- Rothstein R, Michel B, Gangloff S (January 2000). "Replication fork pausing and recombination or "gimme a break"". Genes & Development. 14 (1): 1–10. doi:10.1101/gad.14.1.1. PMID 10640269. S2CID 40751623.
- Bastia D, Zzaman S, Krings G, Saxena M, Peng X, Greenberg MM (September 2008). "Replication termination mechanism as revealed by Tus-mediated polar arrest of a sliding helicase". Proceedings of the National Academy of Sciences of the United States of America. 105 (35): 12831–6. Bibcode:2008PNAS..10512831B. doi:10.1073/pnas.0805898105. PMC 2529109. PMID 18708526.
- Replication factories , PavelHozák, Peter R.Cook, Trends in Cell Biology Volume 4 issue 2; Sciencedirect, DOI
- McGarry TJ, Kirschner MW (June 1998). "Geminin, an inhibitor of DNA replication, is degraded during mitosis". Cell. 93 (6): 1043–53. doi:10.1016/S0092-8674(00)81209-X. PMID 9635433. S2CID 235485.
- de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, Lehmann AR, Hoeijmakers JH (April 2000). "Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice". Current Biology. 10 (8): 479–82. doi:10.1016/S0960-9822(00)00447-4. PMID 10801416. S2CID 15401622.
- Cortez D, Guntuku S, Qin J, Elledge SJ (November 2001). "ATR and ATRIP: partners in checkpoint signaling". Science. 294 (5547): 1713–6. Bibcode:2001Sci...294.1713C. doi:10.1126/science.1065521. PMID 11721054. S2CID 32810119.
- Dart DA, Adams KE, Akerman I, Lakin ND (April 2004). "Recruitment of the cell cycle checkpoint kinase ATR to chromatin during S-phase". The Journal of Biological Chemistry. 279 (16): 16433–40. doi:10.1074/jbc.M314212200. PMID 14871897.
- Ball HL, Myers JS, Cortez D (May 2005). "ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation". Molecular Biology of the Cell. 16 (5): 2372–81. doi:10.1091/mbc.E04-11-1006. PMC 1087242. PMID 15743907.
- Bermudez VP, Lindsey-Boltz LA, Cesare AJ, Maniwa Y, Griffith JD, Hurwitz J, Sancar A (February 2003). "Loading of the human 9-1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro". Proceedings of the National Academy of Sciences of the United States of America. 100 (4): 1633–8. Bibcode:2003PNAS..100.1633B. doi:10.1073/pnas.0437927100. PMC 149884. PMID 12578958.
- Zou L, Cortez D, Elledge SJ (January 2002). "Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin". Genes & Development. 16 (2): 198–208. doi:10.1101/gad.950302. PMC 155323. PMID 11799063.
- Kumagai A, Dunphy WG (October 2000). "Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts". Molecular Cell. 6 (4): 839–49. doi:10.1016/S1097-2765(05)00092-4. PMID 11090622.
- Kumagai A, Dunphy WG (February 2003). "Repeated phosphopeptide motifs in Claspin mediate the regulated binding of Chk1" (PDF). Nature Cell Biology. 5 (2): 161–5. doi:10.1038/ncb921. PMID 12545175. S2CID 29331203.
- Miao H, Seiler JA, Burhans WC (February 2003). "Regulation of cellular and SV40 virus origins of replication by Chk1-dependent intrinsic and UVC radiation-induced checkpoints". The Journal of Biological Chemistry. 278 (6): 4295–304. doi:10.1074/jbc.M204264200. PMID 12424256.
- Bar-Ziv R, Voichek Y, Barkai N (September 2016). "Chromatin dynamics during DNA replication". Genome Research. 26 (9): 1245–56. doi:10.1101/gr.201244.115. PMC 5052047. PMID 27225843.
- Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ (September 1997). "Crystal structure of the nucleosome core particle at 2.8 A resolution". Nature. 389 (6648): 251–60. Bibcode:1997Natur.389..251L. doi:10.1038/38444. PMID 9305837. S2CID 4328827.
- Wittmeyer J, Joss L, Formosa T (July 1999). "Spt16 and Pob3 of Saccharomyces cerevisiae form an essential, abundant heterodimer that is nuclear, chromatin-associated, and copurifies with DNA polymerase alpha". Biochemistry. 38 (28): 8961–71. doi:10.1021/bi982851d. PMID 10413469.
- Wittmeyer J, Formosa T (July 1997). "The Saccharomyces cerevisiae DNA polymerase alpha catalytic subunit interacts with Cdc68/Spt16 and with Pob3, a protein similar to an HMG1-like protein". Molecular and Cellular Biology. 17 (7): 4178–90. doi:10.1128/MCB.17.7.4178. PMC 232271. PMID 9199353.
- Xin H, Takahata S, Blanksma M, McCullough L, Stillman DJ, Formosa T (August 2009). "yFACT induces global accessibility of nucleosomal DNA without H2A-H2B displacement". Molecular Cell. 35 (3): 365–76. doi:10.1016/j.molcel.2009.06.024. PMC 2748400. PMID 19683499.
- Groth A, Corpet A, Cook AJ, Roche D, Bartek J, Lukas J, Almouzni G (December 2007). "Regulation of replication fork progression through histone supply and demand". Science. 318 (5858): 1928–31. Bibcode:2007Sci...318.1928G. doi:10.1126/science.1148992. hdl:1885/53159. PMID 18096807. S2CID 22350778.
- Daganzo SM, Erzberger JP, Lam WM, Skordalakes E, Zhang R, Franco AA, Brill SJ, Adams PD, Berger JM, Kaufman PD (December 2003). "Structure and function of the conserved core of histone deposition protein Asf1". Current Biology. 13 (24): 2148–58. doi:10.1016/j.cub.2003.11.027. PMID 14680630. S2CID 15164132.
- Voichek Y, Bar-Ziv R, Barkai N (March 2016). "Expression homeostasis during DNA replication". Science. 351 (6277): 1087–90. Bibcode:2016Sci...351.1087V. doi:10.1126/science.aad1162. PMID 26941319. S2CID 32751800.
- Stillman B (May 1986). "Chromatin assembly during SV40 DNA replication in vitro". Cell. 45 (4): 555–65. doi:10.1016/0092-8674(86)90287-4. PMID 3011272. S2CID 21212160.
- Shibahara K, Stillman B (February 1999). "Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin". Cell. 96 (4): 575–85. doi:10.1016/S0092-8674(00)80661-3. PMID 10052459. S2CID 13912324.
- Rolef Ben-Shahar T, Castillo AG, Osborne MJ, Borden KL, Kornblatt J, Verreault A (December 2009). "Two fundamentally distinct PCNA interaction peptides contribute to chromatin assembly factor 1 function". Molecular and Cellular Biology. 29 (24): 6353–65. doi:10.1128/MCB.01051-09. PMC 2786881. PMID 19822659.
- Huang S, Zhou H, Katzmann D, Hochstrasser M, Atanasova E, Zhang Z (September 2005). "Rtt106p is a histone chaperone involved in heterochromatin-mediated silencing". Proceedings of the National Academy of Sciences of the United States of America. 102 (38): 13410–5. Bibcode:2005PNAS..10213410H. doi:10.1073/pnas.0506176102. PMC 1224646. PMID 16157874.
- Ito T, Tyler JK, Bulger M, Kobayashi R, Kadonaga JT (October 1996). "ATP-facilitated chromatin assembly with a nucleoplasmin-like protein from Drosophila melanogaster". The Journal of Biological Chemistry. 271 (40): 25041–8. doi:10.1074/jbc.271.40.25041. PMID 8798787.
- Gasser R, Koller T, Sogo JM (May 1996). "The stability of nucleosomes at the replication fork". Journal of Molecular Biology. 258 (2): 224–39. doi:10.1006/jmbi.1996.0245. PMID 8627621.
- Meselson M, Stahl FW (July 1958). "The replication of DNA in Escherichia coli". Proceedings of the National Academy of Sciences of the United States of America. 44 (7): 671–82. Bibcode:1958PNAS...44..671M. doi:10.1073/pnas.44.7.671. PMC 528642. PMID 16590258.
- Bell SP, Kaguni JM (June 2013). "Helicase loading at chromosomal origins of replication". Cold Spring Harbor Perspectives in Biology. 5 (6): a010124. doi:10.1101/cshperspect.a010124. PMC 3660832. PMID 23613349.