Hydrophobic collapse
Hydrophobic collapse is a proposed process for the production of the 3-D conformation adopted by polypeptides and other molecules in polar solvents. The theory states that the nascent polypeptide forms initial secondary structure (ɑ-helices and β-strands) creating localized regions of predominantly hydrophobic residues. The polypeptide interacts with water, thus placing thermodynamic pressures on these regions which then aggregate or "collapse" into a tertiary conformation with a hydrophobic core. Incidentally, polar residues interact favourably with water, thus the solvent-facing surface of the peptide is usually composed of predominantly hydrophilic regions.[1]
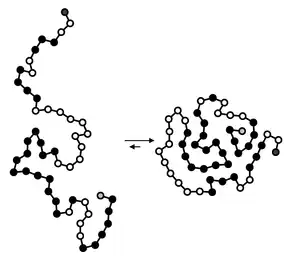
Hydrophobic collapse may also reduce the affinity of conformationally flexible drugs to their protein targets by reducing the net hydrophobic contribution to binding by self association of different parts of the drug while in solution. Conversely rigid scaffolds (also called privileged structures) that resist hydrophobic collapse may enhance drug affinity.[2][3][4]
Partial hydrophobic collapse is an experimentally accepted model for the folding kinetics of many globular proteins, such as myoglobin,[5] alpha-lactalbumin,[6] barstar,[7] and staphylococcal nuclease.[8] However, because experimental evidence of early folding events is difficult to obtain, hydrophobic collapse is often studied in silico via molecular dynamics and Monte Carlo simulations of the folding process.[9][10] Globular proteins that are thought to fold by hydrophobic collapse are particularly amenable to complementary computational and experimental study using phi value analysis.[11]
Biological significance
Correct protein folding is integral to proper functionality within biological systems. Hydrophobic collapse is one of the main events necessary for reaching a protein's stable and functional conformation. Proteins perform extremely specific functions which are dependent on their structure. Proteins that do not fold correctly are nonfunctional and contribute nothing to a biological system.
Hydrophobic aggregation can also occur between unrelated polypeptides. If two locally hydrophobic regions of two unrelated structures are left near each other in aqueous solution, aggregation will occur. In this case, this can have drastic effects on the health of the organism. The formation of amyloid fibrils, insoluble aggregates of hydrophobic protein can lead to a myriad of diseases including Parkinson's and Alzheimer's disease.[12]
Energetics
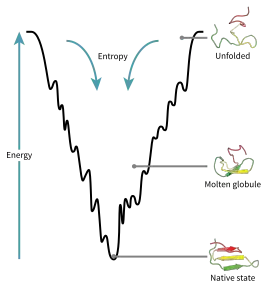
The driving force behind protein folding is not well understood, hydrophobic collapse is a theory, one of many, that is thought to influence how a nascent polypeptide will fold into its native state. Hydrophobic collapse can be visualized as part of the folding funnel model which leads a protein to its lowest kinetically accessible energy state. In this model, we do not consider the interactions of the peptide backbone as this maintains its stability in non-polar and polar environments as long as there is sufficient hydrogen bonding within the backbone, thus we will only consider the thermodynamic contributions of the side chains to protein stability.[13]
When placed in a polar solvent, polar side chains can form weak intermolecular interactions with the solvent, specifically hydrogen bonding. The solvent is able to maintain hydrogen bonding with itself as well as the polypeptide. This maintains the stability of the structure within localized segments of the protein. However, non-polar side chains are unable to participate in hydrogen bonding interactions. The inability of the solvent to interact with these side chains leads to a decrease in entropy of the system. The solvent can interact with itself, however the portion of the molecule in proximity to the non-polar side chain is unable to form any significant interactions, thus the dissociative degrees of freedom available to the molecule decreases and entropy decreases. By aggregating the hydrophobic regions, the solvent can reduce the surface area exposed to non-polar side chains, thus reduce localized areas of decreased entropy. While the entropy of the polypeptide has decreased as it enters a more ordered state, the overall entropy of the system increases, contributing to the thermodynamic favourability of a folded polypeptide.[14]
As can be seen in the folding funnel diagram, the polypeptide is at its highest energy state when unfolded in aqueous solution. As it forms localized folding intermediates, or molten globules, the energy of the system decreases. The polypeptide will continue folding into lower energy states as long as these conformations are kinetically accessible. In this case, a native conformation does not have to be at the lowest energy trough of the diagram as shown, it must simply exist in its natural and kinetically accessible conformation in biological systems.[13]
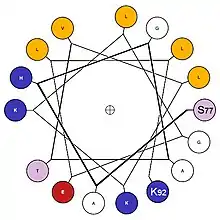
Surface structures

The formation of a hydrophobic core requires the surface structures of this aggregate to maintain contact with both the polar solvent as well as the internal structures. In order to do this, these surface structures usually contain amphipathic properties. A surface exposed alpha helix may have nonpolar residues in an N+3, N+4 position, allowing the alpha-helix to express nonpolar properties on one side when split longitudinally along the axis. Note, in the diagram, the presence of non-polar(gold) amino acids along one side of the helix when viewed through the longitudinal axis, as well as charged/polar amino acids along the other face. This provides this structure with longitudinal amphipathic properties necessary for hydrophobic aggregation along the non-polar side. Similarly, beta strands can also adopt this property with simple alternation of polar and nonpolar residues. Every N+1 side chain will occupy space on the opposite side of the beta strand.[15]
References
- Voet D, Voet JG, Pratt CW (1999). Fundamental of Biochemistry: Life at the Molecular Level (4th ed.). New York: Wiley & Sons, Inc. p. 163. ISBN 978-0470-54784-7.
- Wiley RA, Rich DH (May 1993). "Peptidomimetics derived from natural products". Medicinal Research Reviews. 13 (3): 327–84. doi:10.1002/med.2610130305. PMID 8483337. S2CID 32014299.
- Rich D (1993). "Effect of Hydrophobic Collapse on Enzyme Inhibitor Interactions. Implications for the Design of Peptidomimetics.". In Testa B, Kyburz E, Fuhrer W, Giger R (eds.). Perspectives in Medicinal Chemistry: XIIth International Symposium on Medicinal Chemistry. Weinheim: VCH. pp. 15–25. ISBN 978-3-527-28486-3.
- Rich D, Estiarte M, Hart P (2003). "Stereochemical Aspects of Drug Action I: Conformational Restriction, Steric Hindrance, and Hydrophobic Collapse.". In Wermuth C (ed.). Practice of Medicinal Chemistry (Second ed.). Academic Press. pp. 373–386. doi:10.1016/B978-012744481-9/50027-1. ISBN 978-0-08-049777-8.
- Gilmanshin R, Dyer RB, Callender RH (October 1997). "Structural heterogeneity of the various forms of apomyoglobin: implications for protein folding". Protein Science. 6 (10): 2134–42. doi:10.1002/pro.5560061008. PMC 2143565. PMID 9336836.
- Arai M, Kuwajima K (1996). "Rapid formation of a molten globule intermediate in refolding of alpha-lactalbumin". Folding & Design. 1 (4): 275–87. doi:10.1016/S1359-0278(96)00041-7. PMID 9079390.
- Agashe VR, Shastry MC, Udgaonkar JB (October 1995). "Initial hydrophobic collapse in the folding of barstar". Nature. 377 (6551): 754–7. Bibcode:1995Natur.377..754A. doi:10.1038/377754a0. PMID 7477269. S2CID 4343528.
- Vidugiris GJ, Markley JL, Royer CA (April 1995). "Evidence for a molten globule-like transition state in protein folding from determination of activation volumes". Biochemistry. 34 (15): 4909–12. doi:10.1021/bi00015a001. PMID 7711012.
- Marianayagam NJ, Jackson SE (October 2004). "The folding pathway of ubiquitin from all-atom molecular dynamics simulations". Biophysical Chemistry. 111 (2): 159–71. doi:10.1016/j.bpc.2004.05.009. PMID 15381313.
- Brylinski M, Konieczny L, Roterman I (August 2006). "Hydrophobic collapse in (in silico) protein folding". Computational Biology and Chemistry. 30 (4): 255–67. doi:10.1016/j.compbiolchem.2006.04.007. PMID 16798094.
- Paci E, Friel CT, Lindorff-Larsen K, Radford SE, Karplus M, Vendruscolo M (February 2004). "Comparison of the transition state ensembles for folding of Im7 and Im9 determined using all-atom molecular dynamics simulations with phi value restraints". Proteins. 54 (3): 513–25. doi:10.1002/prot.10595. PMID 14747999. S2CID 490838.
- Stefani M (December 2004). "Protein misfolding and aggregation: new examples in medicine and biology of the dark side of the protein world". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1739 (1): 5–25. doi:10.1016/j.bbadis.2004.08.004. PMID 15607113.
- Govindarajan S, Goldstein RA (May 1998). "On the thermodynamic hypothesis of protein folding". Proceedings of the National Academy of Sciences of the United States of America. 95 (10): 5545–9. Bibcode:1998PNAS...95.5545G. doi:10.1073/pnas.95.10.5545. PMC 20414. PMID 9576919.
- Tanford C (June 1978). "The hydrophobic effect and the organization of living matter". Science. 200 (4345): 1012–8. Bibcode:1978Sci...200.1012T. doi:10.1126/science.653353. JSTOR 1746161. PMID 653353.
- Sharadadevi A, Sivakamasundari C, Nagaraj R (June 2005). "Amphipathic alpha-helices in proteins: results from analysis of protein structures". Proteins. 59 (4): 791–801. doi:10.1002/prot.20459. PMID 15822124. S2CID 85174573.