3-Hydroxy-3-methylglutaryl-CoA lyase
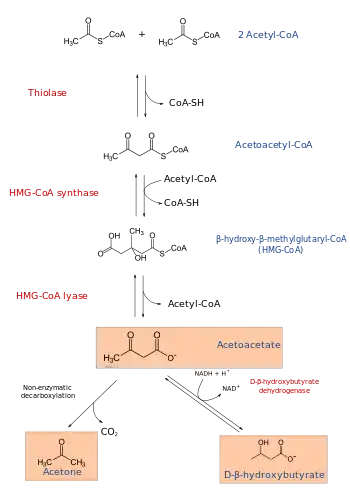
3-Hydroxy-3-methylglutaryl-CoA lyase (or HMG-CoA lyase) is an enzyme (EC 4.1.3.4 that in human is encoded by the HMGCL gene located on chromosome 1. It is a key enzyme in ketogenesis (ketone body formation). It is a ketogenic enzyme in the liver that catalyzes the formation of acetoacetate from HMG-CoA within the mitochondria. It also plays a prominent role in the catabolism of the amino acid leucine.
| 3-hydroxy-3-methylglutaryl-CoA lyase | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Aliases | HMG-CoA_lyasehydroxymethylglutaryl-CoA lyaseHMG-CoA lyase3-hydroxy-3-methylglutaryl coenzyme A lyase(S)-3-hydroxy-3-methylglutaryl-CoA acetoacetate-lyase (acetyl-CoA-forming)(S)-3-hydroxy-3-methylglutaryl-CoA acetoacetate-lyase3-hydroxy-3-methylglutarate-CoA lyasehydroxymethylglutaryl coenzyme A lyase | ||||||||||||||||||||||||||||||||||||||||||||||||||
| External IDs | GeneCards: | ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Hydroxymethylglutaryl-CoA lyase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
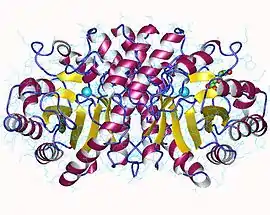 HMG-CoA lyase dimer, Human | |||||||||
| Identifiers | |||||||||
| EC no. | 4.1.3.4 | ||||||||
| CAS no. | 9030-83-5 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
| 3-hydroxymethyl-3-methylglutaryl-Coenzyme A lyase (hydroxymethylglutaricaciduria) | |||||||
|---|---|---|---|---|---|---|---|
| Identifiers | |||||||
| Symbol | HMGCL | ||||||
| NCBI gene | 3155 | ||||||
| HGNC | 5005 | ||||||
| OMIM | 246450 | ||||||
| RefSeq | NM_000191 | ||||||
| UniProt | P35914 | ||||||
| Other data | |||||||
| EC number | 4.1.3.4 | ||||||
| Locus | Chr. 1 p36.1-p35 | ||||||
| |||||||
Structure
The HMGCL gene encodes a 34.5-kDa protein that is localized to the mitochondrion and peroxisome.[1] Multible isoforms of the proteins are known due to alternative splicing. The major isoform (isoform 1) is most highly expressed in the liver[1] whereas isoform 2 is found in energy-demanding tissues including the brain, heart, and skeletal muscle.[2]
Structure of the HMGCL protein has been resolved by X-ray crystallography at 2.1-Å resolution, and reveals that the protein may function as a dimer. Substrate access to the active site of the HMGCL enzyme involves substrate binding across a cavity located at the C-terminal end of a beta barrel structure.[3] In addition, the lysine 48 residue which is mutated in patients with 3-hydroxy-3-methylglutaryl-CoA lyase deficiency is also found to be necessary for substrate binding.[4]
Function
The HMGCL protein plays an essential role in breaking down dietary proteins and fats for energy. It catalyzes the reaction:
- (S)-3-hydroxy-3-methylglutaryl-CoA = acetyl-CoA + acetoacetate
and requires a divalent metal ion as co-factor.[5]
The enzyme is required for ketogenesis in the liver, and is also responsible for processing the amino acid leucine inside the mitochondrion
Deficiency HMG-CoA lyase deficiency causes hypoketotic hypoglycemia similar to that is caused by HMGCS2 mutations but also leads to organic acid accumulation and metabolic acidosis due to altered leucine metabolism. This disorder can be mistaken for Reye syndrome because of the symptoms of vomiting, lethargy, and convulsions.
Clinical significance
Mutations in the HMGCL gene cause 3-hydroxy-3-methylglutaryl-CoA lyase deficiency (HMGCLD), a rare autosomal recessive inborn error of metabolism characterized by disruption of ketogenesis and L-leucine catabolism. To-date more than 30 different mutations including missense mutations of different residues have been associated with patients with HMGCLD in diverse families and ethnicities.[6] HMGCLD typically presents in the first year of the patient's life after a fasting period. Clinical acute symptoms include vomiting, seizures, metabolic acidosis, hypoketotic hypoglycemia, and lethargy.[7]
Interactions
HMGCL interacts with itself to form homodimers and homotetramers. It is also shown in yeast two-hybrid experiments to interact with DNAJA1.
References
- Ashmarina LI, Robert MF, Elsliger MA, Mitchell GA (1996). "Characterization of the hydroxymethylglutaryl-CoA lyase precursor, a protein targeted to peroxisomes and mitochondria". Biochem. J. 315 ( Pt 1): 71–5. doi:10.1042/bj3150071. PMC 1217198. PMID 8670134.
- Puisac B, Ramos M, Arnedo M, Menao S, Gil-Rodríguez MC, Teresa-Rodrigo ME, Pié A, de Karam JC, Wesselink JJ, Giménez I, Ramos FJ, Casals N, Gómez-Puertas P, Hegardt FG, Pié J (2012). "Characterization of splice variants of the genes encoding human mitochondrial HMG-CoA lyase and HMG-CoA synthase, the main enzymes of the ketogenesis pathway". Mol. Biol. Rep. 39 (4): 4777–85. doi:10.1007/s11033-011-1270-8. PMID 21952825. S2CID 16280588.
- Fu Z, Runquist JA, Montgomery C, Miziorko HM, Kim JJ (2010). "Functional insights into human HMG-CoA lyase from structures of Acyl-CoA-containing ternary complexes". J. Biol. Chem. 285 (34): 26341–9. doi:10.1074/jbc.M110.139931. PMC 2924059. PMID 20558737.
- Carrasco P, Menao S, López-Viñas E, Santpere G, Clotet J, Sierra AY, Gratacós E, Puisac B, Gómez-Puertas P, Hegardt FG, Pie J, Casals N (2007). "C-terminal end and aminoacid Lys48 in HMG-CoA lyase are involved in substrate binding and enzyme activity". Mol. Genet. Metab. 91 (2): 120–7. doi:10.1016/j.ymgme.2007.03.007. PMID 17459752.
- Tuinstra RL, Miziorko HM (2003). "Investigation of conserved acidic residues in 3-hydroxy-3-methylglutaryl-CoA lyase: implications for human disease and for functional roles in a family of related proteins". J. Biol. Chem. 278 (39): 37092–8. doi:10.1074/jbc.M304472200. PMID 12874287.
- Menao S, López-Viñas E, Mir C, Puisac B, Gratacós E, Arnedo M, Carrasco P, Moreno S, Ramos M, Gil MC, Pié A, Ribes A, Pérez-Cerda C, Ugarte M, Clayton PT, Korman SH, Serra D, Asins G, Ramos FJ, Gómez-Puertas P, Hegardt FG, Casals N, Pié J (2009). "Ten novel HMGCL mutations in 24 patients of different origin with 3-hydroxy-3-methyl-glutaric aciduria". Human Mutation. 30 (3): E520–9. doi:10.1002/humu.20966. PMID 19177531. S2CID 2826349.
- Online Mendelian Inheritance in Man (OMIM): 3-hydroxy-3-methylglutaryl-coa lyase deficiency; HMGCLD - 246450
External links
- 3-hydroxy-3-methylglutaryl-coenzyme+A+lyase at the U.S. National Library of Medicine Medical Subject Headings (MeSH)