Iodine compounds
Iodine can form compounds using multiple oxidation states. Iodine is quite reactive, but it is much less reactive than the other halogens. For example, while chlorine gas will halogenate carbon monoxide, nitric oxide, and sulfur dioxide (to phosgene, nitrosyl chloride, and sulfuryl chloride respectively), iodine will not do so. Furthermore, iodination of metals tends to result in lower oxidation states than chlorination or bromination; for example, rhenium metal reacts with chlorine to form rhenium hexachloride, but with bromine it forms only rhenium pentabromide and iodine can achieve only rhenium tetraiodide.[2] By the same token, however, since iodine has the lowest ionisation energy among the halogens and is the most easily oxidised of them, it has a more significant cationic chemistry and its higher oxidation states are rather more stable than those of bromine and chlorine, for example in iodine heptafluoride.[1]
| X | XX | HX | BX3 | AlX3 | CX4 |
|---|---|---|---|---|---|
| F | 159 | 574 | 645 | 582 | 456 |
| Cl | 243 | 428 | 444 | 427 | 327 |
| Br | 193 | 363 | 368 | 360 | 272 |
| I | 151 | 294 | 272 | 285 | 239 |
Charge-transfer complexes
The iodine molecule, I2, dissolves in CCl4 and aliphatic hydrocarbons to give bright violet solutions. In these solvents the absorption band maximum occurs in the 520 – 540 nm region and is assigned to a π* to σ* transition. When I2 reacts with Lewis bases in these solvents a blue shift in I2 peak is seen and the new peak (230 – 330 nm) arises that is due to the formation of adducts, which are referred to as charge-transfer complexes.[3]
Hydrogen iodide

The simplest compound of iodine is hydrogen iodide, HI. It is a colourless gas that reacts with oxygen to give water and iodine. Although it is useful in iodination reactions in the laboratory, it does not have large-scale industrial uses, unlike the other hydrogen halides. Commercially, it is usually made by reacting iodine with hydrogen sulfide or hydrazine:[4]
- 2 I2 + N2H4 4 HI + N2
At room temperature, it is a colourless gas, like all of the hydrogen halides except hydrogen fluoride, since hydrogen cannot form strong hydrogen bonds to the large and only mildly electronegative iodine atom. It melts at −51.0 °C and boils at −35.1 °C. It is an endothermic compound that can exothermically dissociate at room temperature, although the process is very slow unless a catalyst is present: the reaction between hydrogen and iodine at room temperature to give hydrogen iodide does not proceed to completion. The H–I bond dissociation energy is likewise the smallest of the hydrogen halides, at 295 kJ/mol.[5]
Aqueous hydrogen iodide is known as hydroiodic acid, which is a strong acid. Hydrogen iodide is exceptionally soluble in water: one litre of water will dissolve 425 litres of hydrogen iodide, and the saturated solution has only four water molecules per molecule of hydrogen iodide.[6] Commercial so-called "concentrated" hydroiodic acid usually contains 48–57% HI by mass; the solution forms an azeotrope with boiling point 126.7 °C at 56.7 g HI per 100 g solution. Hence hydroiodic acid cannot be concentrated past this point by evaporation of water.[5]
Unlike hydrogen fluoride, anhydrous liquid hydrogen iodide is difficult to work with as a solvent, because its boiling point is low, it has a small liquid range, its dielectric constant is low and it does not dissociate appreciably into H2I+ and HI−
2 ions – the latter, in any case, are much less stable than the bifluoride ions (HF−
2) due to the very weak hydrogen bonding between hydrogen and iodine, though its salts with very large and weakly polarising cations such as Cs+ and NR+
4 (R = Me, Et, Bun) may still be isolated. Anhydrous hydrogen iodide is a poor solvent, able to dissolve only small molecular compounds such as nitrosyl chloride and phenol, or salts with very low lattice energies such as tetraalkylammonium halides.[5]
Other binary iodides
Nearly all elements in the periodic table form binary iodides. The exceptions are decidedly in the minority and stem in each case from one of three causes: extreme inertness and reluctance to participate in chemical reactions (the noble gases); extreme nuclear instability hampering chemical investigation before decay and transmutation (many of the heaviest elements beyond bismuth); and having an electronegativity higher than iodine's (oxygen, nitrogen, and the first three halogens), so that the resultant binary compounds are formally not iodides but rather oxides, nitrides, or halides of iodine. (Nonetheless, nitrogen triiodide is named as an iodide as it is analogous to the other nitrogen trihalides.)[7]
Given the large size of the iodide anion and iodine's weak oxidising power, high oxidation states are difficult to achieve in binary iodides, the maximum known being in the pentaiodides of niobium, tantalum, and protactinium. Iodides can be made by reaction of an element or its oxide, hydroxide, or carbonate with hydroiodic acid, and then dehydrated by mildly high temperatures combined with either low pressure or anhydrous hydrogen iodide gas. These methods work best when the iodide product is stable to hydrolysis; otherwise, the possibilities include high-temperature oxidative iodination of the element with iodine or hydrogen iodide, high-temperature iodination of a metal oxide or other halide by iodine, a volatile metal halide, carbon tetraiodide, or an organic iodide. For example, molybdenum(IV) oxide reacts with aluminium(III) iodide at 230 °C to give molybdenum(II) iodide. An example involving halogen exchange is given below, involving the reaction of tantalum(V) chloride with excess aluminium(III) iodide at 400 °C to give tantalum(V) iodide:[7]

Lower iodides may be produced either through thermal decomposition or disproportionation, or by reducing the higher iodide with hydrogen or a metal, for example:[7]
![{\displaystyle {\ce {TaI5{}+Ta->[{\text{thermal gradient}}][{\ce {630^{\circ }C\ ->\ 575^{\circ }C}}]Ta6I14}}}](../I/2bccb303062c4ab95661541d583e04d60a434c25.svg)
Most metal iodides with the metal in low oxidation states (+1 to +3) are ionic. Nonmetals tend to form covalent molecular iodides, as do metals in high oxidation states from +3 and above. Both ionic and covalent iodides are known for metals in oxidation state +3 (e.g. scandium iodide is mostly ionic, but aluminium iodide is not). Ionic iodides MIn tend to have the lowest melting and boiling points among the halides MXn of the same element, because the electrostatic forces of attraction between the cations and anions are weakest for the large iodide anion. In contrast, covalent iodides tend to instead have the highest melting and boiling points among the halides of the same element, since iodine is the most polarisable of the halogens and, having the most electrons among them, can contribute the most to van der Waals forces. Naturally, exceptions abound in intermediate iodides where one trend gives way to the other. Similarly, solubilities in water of predominantly ionic iodides (e.g. potassium and calcium) are the greatest among ionic halides of that element, while those of covalent iodides (e.g. silver) are the lowest of that element. In particular, silver iodide is very insoluble in water and its formation is often used as a qualitative test for iodine.[7]
Iodine halides
The halogens form many binary, diamagnetic interhalogen compounds with stoichiometries XY, XY3, XY5, and XY7 (where X is heavier than Y), and iodine is no exception. Iodine forms all three possible diatomic interhalogens, a trifluoride and trichloride, as well as a pentafluoride and, exceptionally among the halogens, a heptafluoride. Numerous cationic and anionic derivatives are also characterised, such as the wine-red or bright orange compounds of ICl+
2 and the dark brown or purplish black compounds of I2Cl+. Apart from these, some pseudohalides are also known, such as cyanogen iodide (ICN), iodine thiocyanate (ISCN), and iodine azide (IN3).[8]

Iodine monofluoride (IF) is unstable at room temperature and disproportionates very readily and irreversibly to iodine and iodine pentafluoride, and thus cannot be obtained pure. It can be synthesised from the reaction of iodine with fluorine gas in trichlorofluoromethane at −45 °C, with iodine trifluoride in trichlorofluoromethane at −78 °C, or with silver(I) fluoride at 0 °C.[8] Iodine monochloride (ICl) and iodine monobromide (IBr), on the other hand, are moderately stable. The former, a volatile red-brown compound, was discovered independently by Joseph Louis Gay-Lussac and Humphry Davy in 1813–1814 not long after the discoveries of chlorine and iodine, and it mimics the intermediate halogen bromine so well that Justus von Liebig was misled into mistaking bromine (which he had found) for iodine monochloride. Iodine monochloride and iodine monobromide may be prepared simply by reacting iodine with chlorine or bromine at room temperature and purified by fractional crystallisation. Both are quite reactive and attack even platinum and gold, though not boron, carbon, cadmium, lead, zirconium, niobium, molybdenum, and tungsten. Their reaction with organic compounds depends on conditions. Iodine chloride vapour tends to chlorinate phenol and salicyclic acid, since when iodine chloride undergoes homolytic dissociation, chlorine and iodine are produced and the former is more reactive. However, iodine chloride in tetrachloromethane solution results in iodination being the main reaction, since now heterolytic fission of the I–Cl bond occurs and I+ attacks phenol as an electrophile. However, iodine monobromide tends to brominate phenol even in tetrachloromethane solution because it tends to dissociate into its elements in solution, and bromine is more reactive than iodine.[8] When liquid, iodine monochloride and iodine monobromide dissociate into I
2X+
and IX−
2 anions (X = Cl, Br); thus they are significant conductors of electricity and can be used as ionising solvents.[8]
Iodine trifluoride (IF3) is an unstable yellow solid that decomposes above −28 °C. It is thus little-known. It is difficult to produce because fluorine gas would tend to oxidise iodine all the way to the pentafluoride; reaction at low temperature with xenon difluoride is necessary. Iodine trichloride, which exists in the solid state as the planar dimer I2Cl6, is a bright yellow solid, synthesised by reacting iodine with liquid chlorine at −80 °C; caution is necessary during purification because it easily dissociates to iodine monochloride and chlorine and hence can act as a strong chlorinating agent. Liquid iodine trichloride conducts electricity, possibly indicating dissociation to ICl+
2 and ICl−
4 ions.[9]
Iodine pentafluoride (IF5), a colourless, volatile liquid, is the most thermodynamically stable iodine fluoride, and can be made by reacting iodine with fluorine gas at room temperature. It is a fluorinating agent, but is mild enough to store in glass apparatus. Again, slight electrical conductivity is present in the liquid state because of dissociation to IF+
4 and IF−
6. The pentagonal bipyramidal iodine heptafluoride (IF7) is an extremely powerful fluorinating agent, behind only chlorine trifluoride, chlorine pentafluoride, and bromine pentafluoride among the interhalogens: it reacts with almost all the elements even at low temperatures, fluorinates Pyrex glass to form iodine(VII) oxyfluoride (IOF5), and sets carbon monoxide on fire.[10]
Iodine oxides and oxoacids
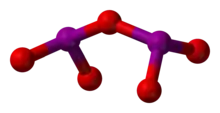
Iodine oxides are the most stable of all the halogen oxides, because of the strong I–O bonds resulting from the large electronegativity difference between iodine and oxygen, and they have been known for the longest time.[11] The stable, white, hygroscopic iodine pentoxide (I2O5) has been known since its formation in 1813 by Gay-Lussac and Davy. It is most easily made by the dehydration of iodic acid (HIO3), of which it is the anhydride. It will quickly oxidise carbon monoxide completely to carbon dioxide at room temperature, and is thus a useful reagent in determining carbon monoxide concentration. It also oxidises nitrogen oxide, ethylene, and hydrogen sulfide. It reacts with sulfur trioxide and peroxydisulfuryl difluoride (S2O6F2) to form salts of the iodyl cation, [IO2]+, and is reduced by concentrated sulfuric acids to iodosyl salts involving [IO]+. It may be fluorinated by fluorine, bromine trifluoride, sulfur tetrafluoride, or chloryl fluoride, resulting iodine pentafluoride, which also reacts with iodine pentoxide, giving iodine(V) oxyfluoride, IOF3. A few other less stable oxides are known, notably I4O9 and I2O4; their structures have not been determined, but reasonable guesses are IIII(IVO3)3 and [IO]+[IO3]− respectively.[12]
| E°(couple) | a(H+) = 1 (acid) | E°(couple) | a(OH−) = 1 (base) |
|---|---|---|---|
| I2/I− | +0.535 | I2/I− | +0.535 |
| HOI/I− | +0.987 | IO−/I− | +0.48 |
| IO− 3/I− | +0.26 | ||
| HOI/I2 | +1.439 | IO−/I2 | +0.42 |
| IO− 3/I2 | +1.195 | ||
| IO− 3/HOI | +1.134 | IO− 3/IO− | +0.15 |
| IO− 4/IO− 3 | +1.653 | ||
| H5IO6/IO− 3 | +1.601 | H 3IO2− 6/IO− 3 | +0.65 |
More important are the four oxoacids: hypoiodous acid (HIO), iodous acid (HIO2), iodic acid (HIO3), and periodic acid (HIO4 or H5IO6). When iodine dissolves in aqueous solution, the following reactions occur:[13]
I2 + H2O ⇌ HIO + H+ + I− Kac = 2.0 × 10−13 mol2 l−2 I2 + 2 OH− ⇌ IO− + H2O + I− Kalk = 30 mol−1 l
Hypoiodous acid is unstable to disproportionation. The hypoiodite ions thus formed disproportionate immediately to give iodide and iodate:[13]
3 IO− ⇌ 2 I− + IO−
3K = 1020
Iodous acid and iodite are even less stable and exist only as a fleeting intermediate in the oxidation of iodide to iodate, if at all.[13] Iodates are by far the most important of these compounds, which can be made by oxidising alkali metal iodides with oxygen at 600 °C and high pressure, or by oxidising iodine with chlorates. Unlike chlorates, which disproportionate very slowly to form chloride and perchlorate, iodates are stable to disproportionation in both acidic and alkaline solutions. From these, salts of most metals can be obtained. Iodic acid is most easily made by oxidation of an aqueous iodine suspension by electrolysis or fuming nitric acid. Iodate has the weakest oxidising power of the halates, but reacts the quickest.[14]
Many periodates are known, including not only the expected tetrahedral IO−
4, but also square-pyramidal IO3−
5, octahedral orthoperiodate IO5−
6, [IO3(OH)3]2−, [I2O8(OH2)]4−, and I
2O4−
9. They are usually made by oxidising alkaline sodium iodate electrochemically (with lead(IV) oxide as the anode) or by chlorine gas:[15]
- IO−
3 + 6 OH− → IO5−
6 + 3 H2O + 2 e− - IO−
3 + 6 OH− + Cl2 → IO5−
6 + 2 Cl− + 3 H2O
They are thermodymically and kinetically powerful oxidising agents, quickly oxidising Mn2+ to MnO−
4, and cleaving glycols, α-diketones, α-ketols, α-aminoalcohols, and α-diamines.[15] Orthoperiodate especially stabilises high oxidation states among metals because of its very high negative charge of −5. Orthoperiodic acid, H5IO6, is stable, and dehydrates at 100 °C in a vacuum to metaperiodic acid, HIO4. Attempting to go further does not result in the nonexistent iodine heptoxide (I2O7), but rather iodine pentoxide and oxygen. Periodic acid may be protonated by sulfuric acid to give the I(OH)+
6 cation, isoelectronic to Te(OH)6 and Sb(OH)−
6, and giving salts with bisulfate and sulfate.[11]
Polyiodine compounds
When iodine dissolves in strong acids, such as fuming sulfuric acid, a bright blue paramagnetic solution including I+
2 cations is formed. A solid salt of the diiodine cation may be obtained by oxidising iodine with antimony pentafluoride:[11]
- 2 I2 + 5 SbF5 2 I2Sb2F11 + SbF3
The salt I2Sb2F11 is dark blue, and the blue tantalum analogue I2Ta2F11 is also known. Whereas the I–I bond length in I2 is 267 pm, that in I+
2 is only 256 pm as the missing electron in the latter has been removed from an antibonding orbital, making the bond stronger and hence shorter. In fluorosulfuric acid solution, deep-blue I+
2 reversibly dimerises below −60 °C, forming red rectangular diamagnetic I2+
4. Other polyiodine cations are not as well-characterised, including bent dark-brown or black I+
3 and centrosymmetric C2h green or black I+
5, known in the AsF−
6 and AlCl−
4 salts among others.[11][16]
The only important polyiodide anion in aqueous solution is linear triiodide, I−
3. Its formation explains why the solubility of iodine in water may be increased by the addition of potassium iodide solution:[11]
- I2 + I− ⇌ I−
3 (Keq = ~700 at 20 °C)
Many other polyiodides may be found when solutions containing iodine and iodide crystallise, such as I−
5, I−
9, I2−
4, and I2−
8, whose salts with large, weakly polarising cations such as Cs+ may be isolated.[11][17]
Organoiodine compounds

Organoiodine compounds have been fundamental in the development of organic synthesis, such as in the Hofmann elimination of amines,[18] the Williamson ether synthesis,[19] the Wurtz coupling reaction,[20] and in Grignard reagents.[21]
The carbon–iodine bond is a common functional group that forms part of core organic chemistry; formally, these compounds may be thought of as organic derivatives of the iodide anion. The simplest organoiodine compounds, alkyl iodides, may be synthesised by the reaction of alcohols with phosphorus triiodide; these may then be used in nucleophilic substitution reactions, or for preparing Grignard reagents. The C–I bond is the weakest of all the carbon–halogen bonds due to the minuscule difference in electronegativity between carbon (2.55) and iodine (2.66). As such, iodide is the best leaving group among the halogens, to such an extent that many organoiodine compounds turn yellow when stored over time due to decomposition into elemental iodine; as such, they are commonly used in organic synthesis, because of the easy formation and cleavage of the C–I bond.[22] They are also significantly denser than the other organohalogen compounds thanks to the high atomic weight of iodine.[23] A few organic oxidising agents like the iodanes contain iodine in a higher oxidation state than −1, such as 2-iodoxybenzoic acid, a common reagent for the oxidation of alcohols to aldehydes,[24] and iodobenzene dichloride (PhICl2), used for the selective chlorination of alkenes and alkynes.[25] One of the more well-known uses of organoiodine compounds is the so-called iodoform test, where iodoform (CHI3) is produced by the exhaustive iodination of a methyl ketone (or another compound capable of being oxidised to a methyl ketone), as follows:[26]

Some drawbacks of using organoiodine compounds as compared to organochlorine or organobromine compounds is the greater expense and toxicity of the iodine derivatives, since iodine is expensive and organoiodine compounds are stronger alkylating agents.[27] For example, iodoacetamide and iodoacetic acid denature proteins by irreversibly alkylating cysteine residues and preventing the reformation of disulfide linkages.[28]
Halogen exchange to produce iodoalkanes by the Finkelstein reaction is slightly complicated by the fact that iodide is a better leaving group than chloride or bromide. The difference is nevertheless small enough that the reaction can be driven to completion by exploiting the differential solubility of halide salts, or by using a large excess of the halide salt.[26] In the classic Finkelstein reaction, an alkyl chloride or an alkyl bromide is converted to an alkyl iodide by treatment with a solution of sodium iodide in acetone. Sodium iodide is soluble in acetone and sodium chloride and sodium bromide are not.[29] The reaction is driven toward products by mass action due to the precipitation of the insoluble salt.[30][31]
See also
References
- Greenwood and Earnshaw, pp. 804-9
- Greenwood and Earnshaw, pp. 800–4
- Greenwood and Earnshaw, pp. 806-7
- Greenwood and Earnshaw, pp. 809–12
- Greenwood and Earnshaw, pp. 812–9
- Holleman, A. F.; Wiberg, E. "Inorganic Chemistry" Academic Press: San Diego, 2001. ISBN 0-12-352651-5.
- Greenwood and Earnshaw, pp. 821–4
- Greenwood and Earnshaw, pp. 824–8
- Greenwood and Earnshaw, pp. 828–831
- Greenwood and Earnshaw, pp. 832–835
- King RB (1995). Inorganic Chemistry of Main Group Elements. Wiley-VCH. pp. 173–98. ISBN 978-0-471-18602-1.
- Greenwood and Earnshaw, pp. 851–3
- Greenwood and Earnshaw, pp. 853–9
- Greenwood and Earnshaw, pp. 863–4
- Greenwood and Earnshaw, pp. 872–5
- Greenwood and Earnshaw, pp. 842–4
- Greenwood and Earnshaw, pp. 835–9
- Hofmann AW (1851). "Beiträge zur Kenntniss der flüchtigen organischen Basen". Annalen der Chemie und Pharmacie. 78 (3): 253–286. doi:10.1002/jlac.18510780302.
- Williamson A (1850). "Theory of Aetherification". Philosophical Magazine. 37 (251): 350–356. doi:10.1080/14786445008646627. (Link to excerpt.)
- Wurtz A (1855). "Ueber eine neue Klasse organischer Radicale". Annalen der Chemie und Pharmacie. 96 (3): 364–375. doi:10.1002/jlac.18550960310.
- Grignard V (1900). "Sur quelques nouvelles combinaisons organométaliques du magnésium et leur application à des synthèses d'alcools et d'hydrocabures". Compt. Rend. 130: 1322–25.
- Lyday PA. "Iodine and Iodine Compounds". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a14_381.
- Blanksby SJ, Ellison GB (April 2003). "Bond dissociation energies of organic molecules" (PDF). Accounts of Chemical Research. 36 (4): 255–263. CiteSeerX 10.1.1.616.3043. doi:10.1021/ar020230d. PMID 12693923. Archived from the original (PDF) on 6 February 2009. Retrieved 25 October 2017.
- Boeckman Jr RK, Shao P, Mullins JJ (2000). "Dess–Martin periodinane: 1,1,1-Triacetoxy-1,1-dihydro-1,2-benziodoxol-3(1H)-one" (PDF). Organic Syntheses. 77: 141.; Collective Volume, vol. 10, p. 696
- Jung ME, Parker MH (October 1997). "Synthesis of Several Naturally Occurring Polyhalogenated Monoterpenes of the Halomon Class(1)". The Journal of Organic Chemistry. 62 (21): 7094–7095. doi:10.1021/jo971371. PMID 11671809.
- Smith, Michael B.; March, Jerry (2007), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (6th ed.), New York: Wiley-Interscience, ISBN 978-0-471-72091-1
- "Safety data for iodomethane". Oxford University.
- Polgár L (August 1979). "Deuterium isotope effects on papain acylation. Evidence for lack of general base catalysis and for enzyme--leaving-group interaction". European Journal of Biochemistry. 98 (2): 369–374. doi:10.1111/j.1432-1033.1979.tb13196.x. PMID 488108.
- Ervithayasuporn V, Ervithayasuporn V, Pornsamutsin N, Pornsamutsin N, Prangyoo P, Prangyoo P, et al. (October 2013). "One-pot synthesis of halogen exchanged silsesquioxanes: octakis(3-bromopropyl)octasilsesquioxane and octakis(3-iodopropyl)octasilsesquioxane". Dalton Transactions. 42 (37): 13747–13753. doi:10.1039/C3DT51373D. PMID 23907310. S2CID 41232118.
- Streitwieser A (1956). "Solvolytic Displacement Reactions at Saturated Carbon Atoms". Chem. Rev. 56 (4): 571–752. doi:10.1021/cr50010a001.
- Bordwell FG, Brannen WT (1964). "The Effect of the Carbonyl and Related Groups on the Reactivity of Halides in SN2 Reactions". J. Am. Chem. Soc. 86 (21): 4645–4650. doi:10.1021/ja01075a025.