Polyurethane
Polyurethane (/ˌpɒliˈjʊərəˌθeɪn, -jʊəˈrɛθeɪn/;[1] often abbreviated PUR and PU) refers to a class of polymers composed of organic units joined by carbamate (urethane) links. In contrast to other common polymers such as polyethylene and polystyrene, polyurethane is produced from a wide range of starting materials. This chemical variety produces polyurethanes with different chemical structures leading to many different applications. These include rigid and flexible foams, and coatings, adhesives, electrical potting compounds, and fibers such as spandex and polyurethane laminate (PUL). Foams are the largest application accounting for 67% of all polyurethane produced in 2016.[2]
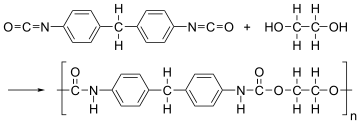

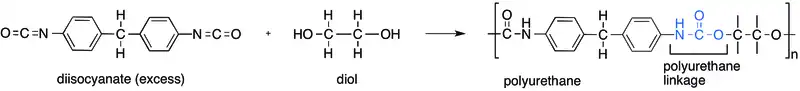
A polyurethane is typically produced by reacting an isocyanate with a polyol.[3] Since a polyurethane contains two types of monomers, which polymerize one after the other, they are classed as alternating copolymers. Both the isocyanates and polyols used to make a polyurethane contain two or more functional groups per molecule.
Global production in 2019 was 25 million metric tonnes,[4] accounting for about 6% of all polymers produced in that year. Polyurethane is a commodity plastic.
History

Otto Bayer and his coworkers at IG Farben in Leverkusen, Germany, first made polyurethanes in 1937.[5][6] The new polymers had some advantages over existing plastics that were made by polymerizing olefins or by polycondensation, and were not covered by patents obtained by Wallace Carothers on polyesters.[7] Early work focused on the production of fibers and flexible foams and PUs were applied on a limited scale as aircraft coating during World War II.[7] Polyisocyanates became commercially available in 1952, and production of flexible polyurethane foam began in 1954 by combining toluene diisocyanate (TDI) and polyester polyols. These materials were also used to produce rigid foams, gum rubber, and elastomers. Linear fibers were produced from hexamethylene diisocyanate (HDI) and 1,4-Butanediol (BDO).
DuPont introduced polyethers, specifically poly(tetramethylene ether) glycol, in 1956. BASF and Dow Chemical introduced polyalkylene glycols in 1957. Polyether polyols were cheaper, easier to handle and more water-resistant than polyester polyols. Union Carbide and Mobay, a U.S. Monsanto/Bayer joint venture, also began making polyurethane chemicals.[7] In 1960 more than 45,000 metric tons of flexible polyurethane foams were produced. The availability of chlorofluoroalkane blowing agents, inexpensive polyether polyols, and methylene diphenyl diisocyanate (MDI) allowed polyurethane rigid foams to be used as high-performance insulation materials. In 1967, urethane-modified polyisocyanurate rigid foams were introduced, offering even better thermal stability and flammability resistance. During the 1960s, automotive interior safety components, such as instrument and door panels, were produced by back-filling thermoplastic skins with semi-rigid foam.
In 1969, Bayer exhibited an all-plastic car in Düsseldorf, Germany. Parts of this car, such as the fascia and body panels, were manufactured using a new process called reaction injection molding (RIM), in which the reactants were mixed and then injected into a mold. The addition of fillers, such as milled glass, mica, and processed mineral fibers, gave rise to reinforced RIM (RRIM), which provided improvements in flexural modulus (stiffness), reduction in coefficient of thermal expansion and better thermal stability. This technology was used to make the first plastic-body automobile in the United States, the Pontiac Fiero, in 1983. Further increases in stiffness were obtained by incorporating pre-placed glass mats into the RIM mold cavity, also known broadly as resin injection molding, or structural RIM.
Starting in the early 1980s, water-blown microcellular flexible foams were used to mold gaskets for automotive panels and air-filter seals, replacing PVC polymers. Polyurethane foams are used in many automotive applications including seating, head and arm rests, and headliners.
Polyurethane foam (including foam rubber) is sometimes made using small amounts of blowing agents to give less dense foam, better cushioning/energy absorption or thermal insulation. In the early 1990s, because of their impact on ozone depletion, the Montreal Protocol restricted the use of many chlorine-containing blowing agents, such as trichlorofluoromethane (CFC-11). By the late 1990s, blowing agents such as carbon dioxide, pentane, 1,1,1,2-tetrafluoroethane (HFC-134a) and 1,1,1,3,3-pentafluoropropane (HFC-245fa) were widely used in North America and the EU, although chlorinated blowing agents remained in use in many developing countries. Later, HFC-134a was also banned due to high ODP and GWP readings, and HFC - 141B was introduced in early 2000s as an alternate blowing agent in developing nations.[8]
Chemistry
Polyurethanes are produced by reacting diisocyanates with polyols,[9][10][11][12][13][14] often in the presence of a catalyst, or upon exposure to ultraviolet radiation.[15] Common catalysts include tertiary amines, such as DABCO, DMDEE, or metallic soaps, such as dibutyltin dilaurate. The stoichiometry of the starting materials must be carefully controlled as excess isocyanate can trimerise, leading to the formation of rigid polyisocyanurates. The polymer usually has a highly crosslinked molecular structure, resulting in a thermosetting material which does not melt on heating; although some thermoplastic polyurethanes are also produced.
The most common application of polyurethane is as solid foams, which requires the presence of a gas, or blowing agent, during the polymerization step. This is commonly achieved by adding small amounts of water, which reacts with isocyanates to form CO2 gas and an amine, via an unstable carbamic acid group. The amine produced can also react with isocyanates to form urea groups, and as such the polymer will contain both these and urethane linkers. The urea is not very soluble in the reaction mixture and tends to form separate "hard segment" phases consisting mostly of polyurea. The concentration and organization of these polyurea phases can have a significant impact on the properties of the foam.[16]
![{\displaystyle {\begin{array}{l}{\ce {{R-N=C=O}+H2O->[{\ce {step}}\ 1]R1-{\underset {| \atop \displaystyle H}{N}}-{\overset {\displaystyle O \atop \|}{C}}-O-H->[{\ce {step}}\ 2][{\ce {-CO2}}]{R-NH2}+{R-N=C=O}->[{\ce {step}}\ 3]-R-{\underset {| \atop \displaystyle H}{N}}-{\overset {\displaystyle O \atop \|}{C}}-{\underset {| \atop \displaystyle H}{N}}-R}}{-}\end{array}}}](../I/5eb505ebc6b4c6a6c75d1183f436772b219512c4.svg)
The type of foam produced can be controlled by regulating the amount of blowing agent and also by the addition of various surfactants which change the rheology of the polymerising mixture. Foams can be either "closed-cell", where most of the original bubbles or cells remain intact, or "open-cell", where the bubbles have broken but the edges of the bubbles are stiff enough to retain their shape, in extreme cases reticulated foams can be formed. Open-cell foams feel soft and allow air to flow through, so they are comfortable when used in seat cushions or mattresses. Closed-cell foams are used as rigid thermal insulation. High-density microcellular foams can be formed without the addition of blowing agents by mechanically frothing the polyol prior to use. These are tough elastomeric materials used in covering car steering wheels or shoe soles.
The properties of a polyurethane are greatly influenced by the types of isocyanates and polyols used to make it. Long, flexible segments, contributed by the polyol, give soft, elastic polymer. High amounts of crosslinking give tough or rigid polymers. Long chains and low crosslinking give a polymer that is very stretchy, short chains with many crosslinks produce a hard polymer while long chains and intermediate crosslinking give a polymer useful for making foam. The choices available for the isocyanates and polyols, in addition to other additives and processing conditions allow polyurethanes to have the very wide range of properties that make them such widely used polymers.
Raw materials
The main ingredients to make a polyurethane are di- and tri-isocyanates and polyols. Other materials are added to aid processing the polymer or to modify the properties of the polymer. PU foam formulation sometimes have water added too.
Isocyanates
Isocyanates used to make polyurethane have two or more isocyanate groups on each molecule. The most commonly used isocyanates are the aromatic diisocyanates, toluene diisocyanate (TDI) and methylene diphenyl diisocyanate, (MDI). These aromatic isocyanates are more reactive than aliphatic isocyanates.
TDI and MDI are generally less expensive and more reactive than other isocyanates. Industrial grade TDI and MDI are mixtures of isomers and MDI often contains polymeric materials. They are used to make flexible foam (for example slabstock foam for mattresses or molded foams for car seats),[17] rigid foam (for example insulating foam in refrigerators) elastomers (shoe soles, for example), and so on. The isocyanates may be modified by partially reacting them with polyols or introducing some other materials to reduce volatility (and hence toxicity) of the isocyanates, decrease their freezing points to make handling easier or to improve the properties of the final polymers.
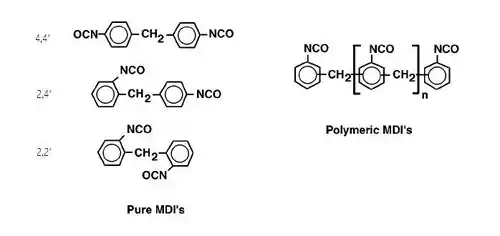
Aliphatic and cycloaliphatic isocyanates are used in smaller quantities, most often in coatings and other applications where color and transparency are important since polyurethanes made with aromatic isocyanates tend to darken on exposure to light.[18] The most important aliphatic and cycloaliphatic isocyanates are 1,6-hexamethylene diisocyanate (HDI), 1-isocyanato-3-isocyanatomethyl-3,5,5-trimethyl-cyclohexane (isophorone diisocyanate, IPDI), and 4,4′-diisocyanato dicyclohexylmethane (H12MDI or hydrogenated MDI). Other more specialized isocyanates include Tetramethylxylylene diisocyanate (TMXDI).
Polyols
Polyols are polymers in their own right and have on average two or more hydroxyl groups per molecule. They can be converted to polyether polyols co-polymerizing ethylene oxide and propylene oxide with a suitable polyol precursor.[19] Polyester polyols are made by the polycondensation of multifunctional carboxylic acids and polyhydroxyl compounds. They can be further classified according to their end use. Higher molecular weight polyols (molecular weights from 2,000 to 10,000) are used to make more flexible polyurethanes while lower molecular weight polyols make more rigid products.
Polyols for flexible applications use low functionality initiators such as dipropylene glycol (f = 2), glycerine (f = 3), or a sorbitol/water solution (f = 2.75).[20] Polyols for rigid applications use high functionality initiators such as sucrose (f = 8), sorbitol (f = 6), toluenediamine (f = 4), and Mannich bases (f = 4). Propylene oxide and/or ethylene oxide is added to the initiators until the desired molecular weight is achieved. The order of addition and the amounts of each oxide affect many polyol properties, such as compatibility, water-solubility, and reactivity. Polyols made with only propylene oxide are terminated with secondary hydroxyl groups and are less reactive than polyols capped with ethylene oxide, which contain primary hydroxyl groups. Incorporating carbon dioxide into the polyol structure is being researched by multiple companies.
Graft polyols (also called filled polyols or polymer polyols) contain finely dispersed styrene–acrylonitrile, acrylonitrile, or polyurea (PHD) polymer solids chemically grafted to a high molecular weight polyether backbone. They are used to increase the load-bearing properties of low-density high-resiliency (HR) foam, as well as add toughness to microcellular foams and cast elastomers. Initiators such as ethylenediamine and triethanolamine are used to make low molecular weight rigid foam polyols that have built-in catalytic activity due to the presence of nitrogen atoms in the backbone. A special class of polyether polyols, poly(tetramethylene ether) glycols, which are made by polymerizing tetrahydrofuran, are used in high performance coating, wetting and elastomer applications.
Conventional polyester polyols are based on virgin raw materials and are manufactured by the direct polyesterification of high-purity diacids and glycols, such as adipic acid and 1,4-butanediol. Polyester polyols are usually more expensive and more viscous than polyether polyols, but they make polyurethanes with better solvent, abrasion, and cut resistance. Other polyester polyols are based on reclaimed raw materials. They are manufactured by transesterification (glycolysis) of recycled poly(ethyleneterephthalate) (PET) or dimethylterephthalate (DMT) distillation bottoms with glycols such as diethylene glycol. These low molecular weight, aromatic polyester polyols are used in rigid foam, and bring low cost and excellent flammability characteristics to polyisocyanurate (PIR) boardstock and polyurethane spray foam insulation.
Specialty polyols include polycarbonate polyols, polycaprolactone polyols, polybutadiene polyols, and polysulfide polyols. The materials are used in elastomer, sealant, and adhesive applications that require superior weatherability, and resistance to chemical and environmental attack. Natural oil polyols derived from castor oil and other vegetable oils are used to make elastomers, flexible bunstock, and flexible molded foam.
Co-polymerizing chlorotrifluoroethylene or tetrafluoroethylene with vinyl ethers containing hydroxyalkyl vinyl ether produces fluorinated (FEVE) polyols. Two-component fluorinated polyurethanes prepared by reacting FEVE fluorinated polyols with polyisocyanate have been used to make ambient cure paints and coatings. Since fluorinated polyurethanes contain a high percentage of fluorine–carbon bonds, which are the strongest bonds among all chemical bonds, fluorinated polyurethanes exhibit resistance to UV, acids, alkali, salts, chemicals, solvents, weathering, corrosion, fungi and microbial attack. These have been used for high performance coatings and paints.[21]
Phosphorus-containing polyols are available that become chemically bonded to the polyurethane matrix for the use as flame retardants. This covalent linkage prevents migration and leaching of the organophosphorus compound.
Bio-derived materials
Interest in sustainable "green" products raised interest in polyols derived from vegetable oils.[22][23][24] Various oils used in the preparation polyols for polyurethanes include soybean, cotton seed, neem seed, and castor. Vegetable oils are functionalized by various ways and modified to polyetheramide, polyethers, alkyds, etc. Renewable sources used to prepare polyols may be dimer fatty acids or fatty acids.[25] Some biobased and isocyanate-free polyurethanes exploit the reaction between polyamines and cyclic carbonates to produce polyhydroxurethanes.[26]
Chain extenders and cross linkers
Chain extenders (f = 2) and cross linkers (f ≥ 3) are low molecular weight hydroxyl and amine terminated compounds that play an important role in the polymer morphology of polyurethane fibers, elastomers, adhesives, and certain integral skin and microcellular foams. The elastomeric properties of these materials are derived from the phase separation of the hard and soft copolymer segments of the polymer, such that the urethane hard segment domains serve as cross-links between the amorphous polyether (or polyester) soft segment domains. This phase separation occurs because the mainly nonpolar, low melting soft segments are incompatible with the polar, high melting hard segments. The soft segments, which are formed from high molecular weight polyols, are mobile and are normally present in coiled formation, while the hard segments, which are formed from the isocyanate and chain extenders, are stiff and immobile. Because the hard segments are covalently coupled to the soft segments, they inhibit plastic flow of the polymer chains, thus creating elastomeric resiliency. Upon mechanical deformation, a portion of the soft segments are stressed by uncoiling, and the hard segments become aligned in the stress direction. This reorientation of the hard segments and consequent powerful hydrogen bonding contributes to high tensile strength, elongation, and tear resistance values.[12][27][28][29][30] The choice of chain extender also determines flexural, heat, and chemical resistance properties. The most important chain extenders are ethylene glycol, 1,4-butanediol (1,4-BDO or BDO), 1,6-hexanediol, cyclohexane dimethanol and hydroquinone bis(2-hydroxyethyl) ether (HQEE). All of these glycols form polyurethanes that phase separate well and form well defined hard segment domains, and are melt processable. They are all suitable for thermoplastic polyurethanes with the exception of ethylene glycol, since its derived bis-phenyl urethane undergoes unfavorable degradation at high hard segment levels.[10] Diethanolamine and triethanolamine are used in flex molded foams to build firmness and add catalytic activity. Diethyltoluenediamine is used extensively in RIM, and in polyurethane and polyurea elastomer formulations.
| Molecule | Mol. mass |
Density (g/cm3) |
Melting pt (°C) |
Boiling pt (°C) |
|---|---|---|---|---|
| Hydroxyl compounds – difunctional molecules | ||||
| Ethylene glycol | 62.1 | 1.110 | −13.4 | 197.4 |
| Diethylene glycol | 106.1 | 1.111 | −8.7 | 245.5 |
| Triethylene glycol | 150.2 | 1.120 | −7.2 | 287.8 |
| Tetraethylene glycol | 194.2 | 1.123 | −9.4 | 325.6 |
| Propylene glycol | 76.1 | 1.032 | Supercools | 187.4 |
| Dipropylene glycol | 134.2 | 1.022 | Supercools | 232.2 |
| Tripropylene glycol | 192.3 | 1.110 | Supercools | 265.1 |
| 1,3-Propanediol | 76.1 | 1.060 | −28 | 210 |
| 1,3-Butanediol | 92.1 | 1.005 | — | 207.5 |
| 1,4-Butanediol | 92.1 | 1.017 | 20.1 | 235 |
| Neopentyl glycol | 104.2 | — | 130 | 206 |
| 1,6-Hexanediol | 118.2 | 1.017 | 43 | 250 |
| 1,4-Cyclohexanedimethanol | — | — | — | — |
| HQEE | — | — | — | — |
| Ethanolamine | 61.1 | 1.018 | 10.3 | 170 |
| Diethanolamine | 105.1 | 1.097 | 28 | 271 |
| Methyldiethanolamine | 119.1 | 1.043 | −21 | 242 |
| Phenyldiethanolamine | 181.2 | — | 58 | 228 |
| Hydroxyl compounds – trifunctional molecules | ||||
| Glycerol | 92.1 | 1.261 | 18.0 | 290 |
| Trimethylolpropane | — | — | — | — |
| 1,2,6-Hexanetriol | — | — | — | — |
| Triethanolamine | 149.2 | 1.124 | 21 | — |
| Hydroxyl compounds – tetrafunctional molecules | ||||
| Pentaerythritol | 136.2 | — | 260.5 | — |
| N,N,N′,N′-Tetrakis (2-hydroxypropyl) ethylenediamine | — | — | — | — |
| Amine compounds – difunctional molecules | ||||
| Diethyltoluenediamine | 178.3 | 1.022 | — | 308 |
| Dimethylthiotoluenediamine | 214.0 | 1.208 | — | — |
Catalysts
Polyurethane catalysts can be classified into two broad categories, basic and acidic amine. Tertiary amine catalysts function by enhancing the nucleophilicity of the diol component. Alkyl tin carboxylates, oxides and mercaptides oxides function as mild Lewis acids in accelerating the formation of polyurethane. As bases, traditional amine catalysts include triethylenediamine (TEDA, also called DABCO, 1,4-diazabicyclo[2.2.2]octane), dimethylcyclohexylamine (DMCHA), dimethylethanolamine (DMEA), and bis-(2-dimethylaminoethyl)ether, a blowing catalyst also called A-99. A typical Lewis acidic catalyst is dibutyltin dilaurate. The process is highly sensitive to the nature of the catalyst and is also known to be autocatalytic.[32]
Factors affecting catalyst selection include balancing three reactions: urethane (polyol+isocyanate, or gel) formation, the urea (water+isocyanate, or "blow") formation, or the isocyanate trimerization reaction (e.g., using potassium acetate, to form isocyanurate rings). A variety of specialized catalysts have been developed.[33][34][35]
Surfactants
Surfactants are used to modify the characteristics of both foam and non-foam polyurethane polymers. They take the form of polydimethylsiloxane-polyoxyalkylene block copolymers, silicone oils, nonylphenol ethoxylates, and other organic compounds. In foams, they are used to emulsify the liquid components, regulate cell size, and stabilize the cell structure to prevent collapse and sub-surface voids.[36] In non-foam applications they are used as air release and antifoaming agents, as wetting agents, and are used to eliminate surface defects such as pin holes, orange peel, and sink marks.
Production
Polyurethanes are produced by mixing two or more liquid streams. The polyol stream contains catalysts, surfactants, blowing agents (when making polyurethane foam insulation) and so on. The two components are referred to as a polyurethane system, or simply a system. The isocyanate is commonly referred to in North America as the 'A-side' or just the 'iso'. The blend of polyols and other additives is commonly referred to as the 'B-side' or as the 'poly'. This mixture might also be called a 'resin' or 'resin blend'. In Europe the meanings for 'A-side' and 'B-side' are reversed. Resin blend additives may include chain extenders, cross linkers, surfactants, flame retardants, blowing agents, pigments, and fillers. Polyurethane can be made in a variety of densities and hardnesses by varying the isocyanate, polyol or additives.
Health and safety
Fully reacted polyurethane polymer is chemically inert.[37] No exposure limits have been established in the U.S. by OSHA (Occupational Safety and Health Administration) or ACGIH (American Conference of Governmental Industrial Hygienists). It is not regulated by OSHA for carcinogenicity.
.jpg.webp)
Polyurethanes are combustible.[38] Decomposition from fire can produce significant amounts of carbon monoxide and hydrogen cyanide, in addition to nitrogen oxides, isocyanates, and other toxic products.[39] Because of the flammability of the material, it has to be treated with flame retardants (at least in case of furniture), almost all of which are considered harmful.[40][41] California later issued Technical Bulletin 117 2013 which allowed most polyurethane foam to pass flammability tests without the use of flame retardants. Green Science Policy Institute states: "Although the new standard can be met without flame retardants, it does NOT ban their use. Consumers who wish to reduce household exposure to flame retardants can look for a TB117-2013 tag on furniture, and verify with retailers that products do not contain flame retardants."[42]
Liquid resin blends and isocyanates may contain hazardous or regulated components. Isocyanates are known skin and respiratory sensitizers. Additionally, amines, glycols, and phosphate present in spray polyurethane foams present risks.[43]
Exposure to chemicals that may be emitted during or after application of polyurethane spray foam (such as isocyanates) are harmful to human health and therefore special precautions are required during and after this process.[44]
In the United States, additional health and safety information can be found through organizations such as the Polyurethane Manufacturers Association (PMA) and the Center for the Polyurethanes Industry (CPI), as well as from polyurethane system and raw material manufacturers. Regulatory information can be found in the Code of Federal Regulations Title 21 (Food and Drugs) and Title 40 (Protection of the Environment). In Europe, health and safety information is available from ISOPA,[45] the European Diisocyanate and Polyol Producers Association.
Manufacturing
The methods of manufacturing polyurethane finished goods range from small, hand pour piece-part operations to large, high-volume bunstock and boardstock production lines. Regardless of the end-product, the manufacturing principle is the same: to meter the liquid isocyanate and resin blend at a specified stoichiometric ratio, mix them together until a homogeneous blend is obtained, dispense the reacting liquid into a mold or on to a surface, wait until it cures, then demold the finished part.
Dispensing equipment
Although the capital outlay can be high, it is desirable to use a meter-mix or dispense unit for even low-volume production operations that require a steady output of finished parts. Dispense equipment consists of material holding (day) tanks, metering pumps, a mix head, and a control unit. Often, a conditioning or heater–chiller unit is added to control material temperature in order to improve mix efficiency, cure rate, and to reduce process variability. Choice of dispense equipment components depends on shot size, throughput, material characteristics such as viscosity and filler content, and process control. Material day tanks may be single to hundreds of gallons in size and may be supplied directly from drums, IBCs (intermediate bulk containers, such as caged IBC totes), or bulk storage tanks. They may incorporate level sensors, conditioning jackets, and mixers. Pumps can be sized to meter in single grams per second up to hundreds of pounds per minute. They can be rotary, gear, or piston pumps, or can be specially hardened lance pumps to meter liquids containing highly abrasive fillers such as chopped or hammer-milled glass fiber and wollastonite.
 A high-pressure polyurethane dispense unit, showing control panel, high-pressure pump, integral day tanks, and hydraulic drive unit
A high-pressure polyurethane dispense unit, showing control panel, high-pressure pump, integral day tanks, and hydraulic drive unit A high-pressure mix head, showing simple controls (front view)
A high-pressure mix head, showing simple controls (front view) A high-pressure mix head, showing material supply and hydraulic actuator lines (rear view)
A high-pressure mix head, showing material supply and hydraulic actuator lines (rear view)
The pumps can drive low-pressure (10 to 30 bar, 1 to 3 MPa) or high-pressure (125 to 250 bar, 12.5 to 25.0 MPa) dispense systems. Mix heads can be simple static mix tubes, rotary-element mixers, low-pressure dynamic mixers, or high-pressure hydraulically actuated direct impingement mixers. Control units may have basic on/off and dispense/stop switches, and analogue pressure and temperature gauges, or may be computer-controlled with flow meters to electronically calibrate mix ratio, digital temperature and level sensors, and a full suite of statistical process control software. Add-ons to dispense equipment include nucleation or gas injection units, and third or fourth stream capability for adding pigments or metering in supplemental additive packages.
 A low-pressure mix head with calibration chamber installed, showing material supply and air actuator lines
A low-pressure mix head with calibration chamber installed, showing material supply and air actuator lines Low-pressure mix head components, including mix chambers, conical mixers, and mounting plates
Low-pressure mix head components, including mix chambers, conical mixers, and mounting plates 5-gallon (20-liter) material day tanks for supplying a low-pressure dispense unit
5-gallon (20-liter) material day tanks for supplying a low-pressure dispense unit
Tooling
Distinct from pour-in-place, bun and boardstock, and coating applications, the production of piece parts requires tooling to contain and form the reacting liquid. The choice of mold-making material is dependent on the expected number of uses to end-of-life (EOL), molding pressure, flexibility, and heat transfer characteristics.
RTV silicone is used for tooling that has an EOL in the thousands of parts. It is typically used for molding rigid foam parts, where the ability to stretch and peel the mold around undercuts is needed. The heat transfer characteristic of RTV silicone tooling is poor. High-performance, flexible polyurethane elastomers are also used in this way.
Epoxy, metal-filled epoxy, and metal-coated epoxy is used for tooling that has an EOL in the tens of thousands of parts. It is typically used for molding flexible foam cushions and seating, integral skin and microcellular foam padding, and shallow-draft RIM bezels and fascia. The heat transfer characteristic of epoxy tooling is fair; the heat transfer characteristic of metal-filled and metal-coated epoxy is good. Copper tubing can be incorporated into the body of the tool, allowing hot water to circulate and heat the mold surface.
Aluminum is used for tooling that has an EOL in the hundreds of thousands of parts. It is typically used for molding microcellular foam gasketing and cast elastomer parts, and is milled or extruded into shape.
Mirror-finish stainless steel is used for tooling that imparts a glossy appearance to the finished part. The heat transfer characteristic of metal tooling is excellent.
Finally, molded or milled polypropylene is used to create low-volume tooling for molded gasket applications. Instead of many expensive metal molds, low-cost plastic tooling can be formed from a single metal master, which also allows greater design flexibility. The heat transfer characteristic of polypropylene tooling is poor, which must be taken into consideration during the formulation process.
Applications
In 2007, the global consumption of polyurethane raw materials was above 12 million metric tons, and the average annual growth rate was about 5%.[46] Revenues generated with PUR on the global market are expected to rise to approximately US$75 billion by 2022.[47]
Degradation and environmental fate
Effects of visible light

Polyurethanes, especially those made using aromatic isocyanates, contain chromophores that interact with light. This is of particular interest in the area of polyurethane coatings, where light stability is a critical factor and is the main reason that aliphatic isocyanates are used in making polyurethane coatings. When PU foam, which is made using aromatic isocyanates, is exposed to visible light, it discolors, turning from off-white to yellow to reddish brown. It has been generally accepted that apart from yellowing, visible light has little effect on foam properties.[48][49] This is especially the case if the yellowing happens on the outer portions of a large foam, as the deterioration of properties in the outer portion has little effect on the overall bulk properties of the foam itself.
It has been reported that exposure to visible light can affect the variability of some physical property test results.[50]
Higher-energy UV radiation promotes chemical reactions in foam, some of which are detrimental to the foam structure.[51]
Hydrolysis and biodegradation
Polyurethanes may degrade due to hydrolysis. This is a common problem with shoes left in a closet, and reacting with moisture in the air.[52]
Microbial degradation of polyurethane is believed to be due to the action of esterase, urethanase, hydrolase and protease enzymes.[53] The process is slow as most microbes have difficulty moving beyond the surface of the polymer. Susceptibility to fungi is higher due to their release of extracellular enzymes, which are better able to permeate the polymer matrix. Two species of the Ecuadorian fungus Pestalotiopsis are capable of biodegrading polyurethane in aerobic and anaerobic conditions such as found at the bottom of landfills.[54][55] Degradation of polyurethane items at museums has been reported.[56] Polyester-type polyurethanes are more easily biodegraded by fungus than polyether-type.[57]
See also
References
- "polyurethane". Dictionary.com Unabridged (Online). n.d.
- Gama, Nuno; Ferreira, Artur; Barros-Timmons, Ana (27 September 2018). "Polyurethane Foams: Past, Present, and Future". Materials. 11 (10): 1841. Bibcode:2018Mate...11.1841G. doi:10.3390/ma11101841. PMC 6213201. PMID 30262722.
- "Polyurethane". American Chemistry Council. Retrieved 2022-09-19.
- "Polyurethane global market volume 2015-2026". Statista. Retrieved 23 July 2021.
- Bayer, Otto (1947). "Das Di-Isocyanat-Polyadditionsverfahren (Polyurethane)". Angewandte Chemie. 59 (9): 257–72. Bibcode:1947AngCh..59..257B. doi:10.1002/ange.19470590901.
- DE 728981, I.G. Farbenindustrie A.G., "Verfahren zur Herstellung von Polyurethanen bzw. Polyharnstoffen [Process for the production of polyurethanes or polyurea]", published 1942-12-07
- Seymour, Raymond B.; Kauffman, George B. (1992). "Polyurethanes: A class of modern versatile materials". Journal of Chemical Education. 69 (11): 909. Bibcode:1992JChEd..69..909S. doi:10.1021/ed069p909.
- Feske, Bert (October 2004). "The Use of Saytex RB-9130/9170 Low Viscosity Brominated Flame Retardant Polyols in HFC-245fa and High Water Formulations" (PDF). Polyurethanes Expo 2004. Las Vegas, NV: Alliance for the Polyurethane Industry Technical Conference. p. 309. Retrieved 2007-08-01.
- n ≥ 2
- Gum, Wilson; Riese, Wolfram; Ulrich, Henri (1992). Reaction Polymers. New York: Oxford University Press. ISBN 978-0-19-520933-4.
- Harrington, Ron; Hock, Kathy (1991). Flexible Polyurethane Foams. Midland: The Dow Chemical Company.
- Oertel, Gunter (1985). Polyurethane Handbook. New York: Macmillen Publishing Co., Inc. ISBN 978-0-02-948920-8.
- Ulrich, Henri (1996). Chemistry and Technology of Isocyanates. New York: John Wiley & Sons, Inc. ISBN 978-0-471-96371-4.
- Woods, George (1990). The ICI Polyurethanes Book. New York: John Wiley & Sons, Inc. ISBN 978-0-471-92658-0.
- Soto, Marc; Sebastián, Rosa María; Marquet, Jordi (2014). "Photochemical Activation of Extremely Weak Nucleophiles: Highly Fluorinated Urethanes and Polyurethanes from Polyfluoro Alcohols". The Journal of Organic Chemistry. 79 (11): 5019–27. doi:10.1021/jo5005789. PMID 24820955.
- Kaushiva, Byran D. (August 15, 1999). Structure-Property Relationships of Flexible Polyurethane Foams (Ph.D.). Virginia Polytechnic Institute.
- "Technical data sheet from Dow Chemical". Archived from the original on 2007-10-13. Retrieved 2007-09-15.
- Randall, David; Lee, Steve (2002). The Polyurethanes Book. New York: Wiley. ISBN 978-0-470-85041-1.
- Petrović, Zoran S. (2008). "Polyurethanes from Vegetable Oils". Polymer Reviews. 48 (1): 109–155. doi:10.1080/15583720701834224. S2CID 95466690.
- EP 0755955, Hager, Stanley L.; Knight, James E. & Helma, Gregory F. et al., "Polyether polyols suitable for flexible polyurethane foam prepared by co-initiation of aqueous solutions of solid polyhydroxyl initiators", published 1997-01-29, assigned to ARCO Chemical Technology
- Bob Parker. "FEVE Technology for Higher Performance Coating Systems on Bridges" (PDF). Paintsquare.com. Retrieved 5 March 2022.
- Khanderay, Jitendra C., and Vikas V. Gite. "Vegetable oil-based polyurethane coatings: recent developments in India." Green Materials 5.3 (2017): 109-122.
- Niemeyer, Timothy; Patel, Munjal; Geiger, Eric (September 2006). A Further Examination of Soy-Based Polyols in Polyurethane Systems. Salt Lake City, UT: Alliance for the Polyurethane Industry Technical Conference.
- "New Twist on Green: 2008 Ford Mustang Seats Will Be Soy-Based Foam". Edmunds inside line. July 12, 2007. Archived from the original on 2008-05-31. Retrieved 2010-06-15.
- Biobased dimer fatty acid containing two pack polyurethane for wood finished coatings, SD Rajput, PP Mahulikar, VV Gite, Progress in Organic Coatings 77 (1), 38-46
- Nohra, Bassam; Candy, Laure; Blanco, Jean-François; Guerin, Celine; Raoul, Yann; Mouloungui, Zephirin (2013). "From Petrochemical Polyurethanes to Biobased Polyhydroxyurethanes" (PDF). Macromolecules. 46 (10): 3771–92. Bibcode:2013MaMol..46.3771N. doi:10.1021/ma400197c. Archived (PDF) from the original on 2017-09-22.
- Blackwell, J.; Nagarajan, M. R.; Hoitink, T. B. (1981). The Structure of the Hard Segments in MDI/diol/PTMA Polyurethane Elastomers. pp. 179–196. doi:10.1021/bk-1981-0172.ch014. ISBN 978-0-8412-0664-9. ISSN 0097-6156.
{{cite book}}:|journal=ignored (help) - Blackwell, John; Gardner, Kenncorwin H. (1979). "Structure of the hard segments in polyurethane elastomers". Polymer. 20: 13–17. doi:10.1016/0032-3861(79)90035-1. ISSN 0032-3861.
- Grillo, D. J.; Housel, T. L. (1992). "Physical Properties of Polyurethanes from Polyesters and Other Polyols". Polyurethanes '92 Conference Proceedings. New Orleans, LA: The Society of the Plastics Industry, Inc.
- Musselman, S. G.; Santosusso, T. M.; Sperling, L. H. (1998). "Structure Versus Performance Properties of Cast Elastomers". Polyurethanes '98 Conference Proceedings. Dallas, TX: The Society of the Plastics Industry, Inc.
- A Guide to Glycols. Midland, Mich.: The Dow Chemical Co., Chemicals and Metals Department. 1992. Brochure 117-00991-92Hyc.
- Adam, Norbert; Avar, Geza; Blankenheim, Herbert; Friederichs, Wolfgang; Giersig, Manfred; Weigand, Eckehard; Halfmann, Michael; Wittbecker, Friedrich-Wilhelm; Larimer (2005). "Polyurethanes". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a21_665.pub2.
- "Jeffcat Amine Catalysts for the Polyurethane Industry" (PDF). 2006. Archived from the original (PDF) on 2007-11-29. Retrieved 2007-10-23.
- "Building quality with Air Products trimerisation catalysts" (PDF). 2003. Archived from the original (PDF) on 2007-11-29. Retrieved 2007-10-23.
- "FOMREZ Specialty Tin Catalysts for Polyurethane Applications". 120-074-10. January 2001.
- Randall, David; Lee, Steve, eds. (2002). "10". The Polyurethanes Book. The United Kingdom: Huntsman International LLC, Polyurethanes business. pp. 156–159. ISBN 978-0470850411.
- Dernehl, C. U. (1966). "Health hazards associated with polyurethane foams". Journal of Occupational Medicine. 8 (2): 59–62. PMID 5903304.
- "Health Alert: Polyurethane exposure" (PDF). Archived from the original (PDF) on 2004-10-19. Retrieved 2009-12-19.
- McKenna, Sean Thomas; Hull, Terence Richard (2016). "The fire toxicity of polyurethane foams". Fire Science Reviews. 5: 3. doi:10.1186/s40038-016-0012-3.
- "Environmental Profiles of Chemical Flame-Retardant Alternatives for Low-Density Polyurethane Foam". United States Environmental Protection Agency. September 2005.
- "Flame Retardants Used in Flexible Polyurethane Foam – Draft Update to a 2005 Alternatives Assessment". United States Environmental Protection Agency. June 2014.
- "Manufacturers" (PDF). greensciencepolicy.org. 2015. Archived (PDF) from the original on 2015-12-19.
- "Help Wanted: Spray Polyurethane Foam Insulation Research". NIOSH Science Blog. CDC.
- "Quick Safety Tips for Spray Polyurethane Foam Users". United States Environmental Protection Agency. 4 August 2015.
- "Home : ISOPA". www.isopa.org.
- Avar, G. (October 2008). "Polyurethanes (PU)". Kunststoffe International (10/2008): 123–7.
- "Market Study: Polyurethanes and Isocyanates". Ceresana. July 2013.
- "Discoloration of polyurethane foam" (PDF). Foamex Information sheet. Archived from the original (PDF) on 2010-09-24. Retrieved 2010-09-26.
- Valentine, C.; Craig, T.A.; Hager, S.L. (1993). "Inhibition of the Discoloration of Polyurethane Foam Caused by Ultraviolet Light". Journal of Cellular Plastics. 29 (6): 569–88. doi:10.1177/0021955X9302900605. S2CID 208363195.
- Blair, G. Ron; Dawe, Bob; McEvoy, Jim; Pask, Roy; de Priamus, Marcela Rusan; Wright, Carol (2007). The Effect of Visible Light on the Variability of Flexible Foam Compression Sets (PDF). Orlando, FL: Center for the Polyurethane Industry. Retrieved 2008-01-26.
- Newman, Christopher R.; Forciniti, Daniel (2001). "Modeling the Ultraviolet Photodegradation of Rigid Polyurethane Foams". Industrial & Engineering Chemistry Research. 40 (15): 3346–52. doi:10.1021/ie0009738.
- "Hydrolysis, The Crumbling of Shoe Soles explained | Safety Shoes and Gloves". www.safetyjogger.com.
- Toward, Gary T. (June 2002). "Biodegradation of polyurethane: a review". International Biodeterioration & Biodegradation. 49 (4): 245–252. doi:10.1016/S0964-8305(02)00051-3.
- Russell, J. R.; Huang, J.; Anand, P.; Kucera, K.; Sandoval, A. G.; Dantzler, K. W.; Hickman, D.; Jee, J.; Kimovec, F. M.; Koppstein, D.; Marks, D. H.; Mittermiller, P. A.; Nunez, S. J.; Santiago, M.; Townes, M. A.; Vishnevetsky, M.; Williams, N. E.; Vargas, M. P. N.; Boulanger, L.-A.; Bascom-Slack, C.; Strobel, S. A. (2011). "Biodegradation of Polyester Polyurethane by Endophytic Fungi". Applied and Environmental Microbiology. 77 (17): 6076–84. Bibcode:2011ApEnM..77.6076R. doi:10.1128/AEM.00521-11. PMC 3165411. PMID 21764951.
- "Could Plastic-Eating Mushrooms Solve mankind's Plastic Problem?". Sciencemint. 2021-04-14. Retrieved 2021-07-02.
- Cappitelli, F.; Sorlini, C. (2007). "Microorganisms Attack Synthetic Polymers in Items Representing Our Cultural Heritage". Applied and Environmental Microbiology. 74 (3): 564–9. doi:10.1128/AEM.01768-07. PMC 2227722. PMID 18065627.
- Tokiwa, Yutaka; Calabia, Buenaventurada P.; Ugwu, Charles U.; Aiba, Seiichi (2009). "Biodegradability of Plastics". International Journal of Molecular Sciences. 10 (9): 3722–42. doi:10.3390/ijms10093722. PMC 2769161. PMID 19865515.
External links
- Center for the Polyurethanes Industry: information for EH&S issues related to polyurethanes developments
- Polyurethane synthesis, Polymer Science Learning Center, University of Southern Mississippi
- Polyurethane Foam Association: Industry information, educational materials and resources related to flexible polyurethane foam
- PU Europe: European PU insulation industry association (formerly BING): European voice for the national trade associations representing the polyurethane insulation industry
- ISOPA: European Diisocyanate & Polyol Producers Association: ISOPA represents the manufacturers in Europe of aromatic diisocyanates and polyols

