Non-coding DNA
Non-coding DNA (ncDNA) sequences are components of an organism's DNA that do not encode protein sequences. Some non-coding DNA is transcribed into functional non-coding RNA molecules (e.g. transfer RNA, microRNA, piRNA, ribosomal RNA, and regulatory RNAs). Other functional regions of the non-coding DNA fraction include regulatory sequences that control gene expression; scaffold attachment regions; origins of DNA replication; centromeres; and telomeres. Some non-coding regions appear to be mostly nonfunctional such as introns, pseudogenes, intergenic DNA, and fragments of transposons and viruses.
Fraction of non-coding genomic DNA
In bacteria, the coding regions typically take up 88% of the genome.[1] The remaining 12% does not encode proteins, but much of it still has biological function through genes where the RNA transcript is functional (non-coding genes) and regulatory sequences, which means that almost all of the bacterial genome has a function.[1] The amount of coding DNA in eukaryrotes is usually a much smaller fraction of the genome because eukaryotic genomes contain large amounts of repetitive DNA not found in prokaryotes. The human genome contains somewhere between 1–2% coding DNA.[2][3] The exact number is not known because there are disputes over the number of functional coding exons and over the total size of the human genome. This means that 98–99% of the human genome consists of non-coding DNA and this includes many functional elements such as non-coding genes and regulatory sequences.
Genome size in eukaryotes can vary over a wide range, even between closely related species. This puzzling observation was originally known as the C-value Paradox where "C" refers to the haploid genome size.[4] The paradox was resolved with the discovery that most of the differences were due to the expansion and contraction of repetitive DNA and not the number of genes. Some researchers speculated that this repetitive DNA was mostly junk DNA. The reasons for the changes in genome size are still being worked out and this problem is called the C-value Enigma.[5]
This led to the observation that the number of genes does not seem to correlate with perceived notions of complexity because the number of genes seems to be relatively constant, an issue termed the G-value Paradox.[6] For example, the genome of the unicellular Polychaos dubium (formerly known as Amoeba dubia) has been reported to contain more than 200 times the amount of DNA in humans (i.e. more than 600 billion pairs of bases vs a bit more than 3 billion in humans).[7] The pufferfish Takifugu rubripes genome is only about one eighth the size of the human genome, yet seems to have a comparable number of genes. Genes take up about 30% of the pufferfish genome and the coding DNA is about 10%. (Non-coding DNA = 90%.) The reduced size of the pufferfish genome is due to a reduction in the length of introns and less repetitive DNA.[8][9]
Utricularia gibba, a bladderwort plant, has a very small nuclear genome (100.7 Mb) compared to most plants.[10][11] It likely evolved from an ancestral genome that was 1,500 Mb in size.[11] The bladderwort genome has roughly the same number of genes as other plants but the total amount of coding DNA comes to about 30% of the genome.[10][11]
The remainder of the genome (70% non-coding DNA) consists of promoters and regulatory sequences that are shorter than those in other plant species.[10] The genes contain introns but there are fewer of them and they are smaller than the introns in other plant genomes.[10] There are noncoding genes, including many copies of ribosomal RNA genes.[11] The genome also contains telomere sequences and centromeres as expected.[11] Much of the repetitive DNA seen in other eukaryotes has been deleted from the bladderwort genome since that lineage split from those of other plants. About 59% of the bladderwort genome consists of transposon-related sequences but since the genome is so much smaller than other genomes, this represents a considerable reduction in the amount of this DNA.[11] The authors of the original 2013 article note that claims of additional functional elements in the non-coding DNA of animals do not seem to apply to plant genomes.[10]
According to a New York Times article, during the evolution of this species, "... genetic junk that didn't serve a purpose was expunged, and the necessary stuff was kept."[12] According to Victor Albert of the University of Buffalo, the plant is able to expunge its so-called junk DNA and "have a perfectly good multicellular plant with lots of different cells, organs, tissue types and flowers, and you can do it without the junk. Junk is not needed."[13]
Types of non-coding DNA sequences
Noncoding genes
There are two types of genes: protein coding genes and noncoding genes.[14] Noncoding genes are an important part of non-coding DNA and they include genes for transfer RNA and ribosomal RNA. These genes were discovered in the 1960s. Prokaryotic genomes contain genes for a number of other noncoding RNAs but noncoding RNA genes are much more common in eukaryotes.
Typical classes of noncoding genes in eukaryotes include genes for small nuclear RNAs (snRNAs), small nucleolar RNAs (sno RNAs), microRNAs (miRNAs), short interfering RNAs (siRNAs), PIWI-interacting RNAs (piRNAs), and long noncoding RNAs (lncRNAs). In addition, there are a number of unique RNA genes that produce catalytic RNAs.[15]
Noncoding genes account for only a few percent of prokaryotic genomes[16] but they can represent a vastly higher fraction in eukaryotic genomes.[17] In humans, the noncoding genes take up at least 6% of the genome, largely because there are hundreds of copies of ribosomal RNA genes. Protein-coding genes occupy about 38% of the genome; a fraction that is much higher than the coding region because genes contain large introns.
The total number of noncoding genes in the human genome is controversial. Some scientists think that there are only about 5,000 noncoding genes while others believe that there may be more than 100,000 (see the article on Non-coding RNA). The difference is largely due to debate over the number of lncRNA genes.[18]
Promoters and regulatory elements
Promoters are DNA segments near the 5' end of the gene where transcription begins. They are the sites where RNA polymerase binds to initiate RNA synthesis. Every gene has a noncoding promoter.
Regulatory elements are sites that control the transcription of a nearby gene. They are almost always sequences where transcription factors bind to DNA and these transcription factors can either activate transcription (activators) or repress transcription (repressors). Regulatory elements were discovered in the 1960s and their general characteristics were worked out in the 1970s by studying specific transcription factors in bacteria and bacteriophage.
Promoters and regulatory sequences represent an abundant class of noncoding DNA but they mostly consist of a collection of relatively short sequences so they don't take up a very large fraction of the genome. The exact amount of regulatory DNA in mammalian genome is unclear because it is difficult to distinguish between spurious transcription factor binding sites and those that are functional. The binding characteristics of typical DNA-binding proteins were characterized in the 1970s and the biochemical properties of transcription factors predict that in cells with large genomes the majority of binding sites will be fortuitous and not biologiacally functional.
Many regulatory sequences occur near promoters, usually upstream of the transcription start site of the gene. Some occur within a gene and a few are located downstream of the transcription termination site. In eukaryotes, there are some regulatory sequences that are located at a considerable distance from the promoter region. These distant regulatory sequences are often called enhancers but there is no rigorous definition of enhancer that distinguishes it from other transcription factor binding sites.[19][20]
Introns
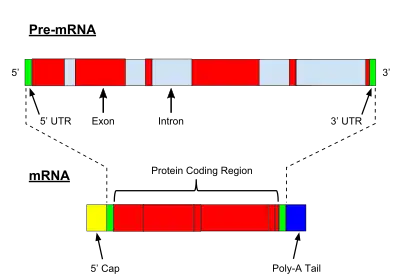
Introns are the parts of a gene that are transcribed into the precursor RNA sequence, but ultimately removed by RNA splicing during the processing to mature RNA. Introns are found in both types of genes: protein-coding genes and noncoding genes. They are present in prokaryotes but they are much more common in eukaryotic genomes.
Group I and group II introns take up only a small percentage of the genome when they are present. Spliceosomal introns (see Figure) are only found in eukaryotes and they can represent a substantial proportion of the genome. In humans, for example, introns in protein-coding genes cover 37% of the genome. Combining that with about 1% coding sequences means that protein-coding genes occupy about 38% of the human genome. The calculations for noncoding genes are more complicated because there's considerable dispute over the total number of noncoding genes but taking only the well-defined examples means that noncoding genes occupy at least 6% of the genome.[21][2]
Untranslated regions
The standard biochemistry and molecular biology textbooks describe non-coding nucleotides in mRNA located between the 5' end of the gene and the translation initiation codon. These regions are called 5'-untranslated regions or 5'-UTRs. Similar regions called 3'-untranslated regions (3'-UTRs) are found at the end of the gene. The 5'-UTRs and 3'UTRs are very short in bacteria but they can be several hundred nucleotides in length in eukaryotes. They contain short elements that control the initiation of translation (5'-UTRs) and transcription termination (3'-UTRs) as well as regulatory elements that may control mRNA stability, processing, and targeting to different regions of the cell.[22][23][24]
Origins of replication
DNA synthesis begins at specific sites called origins of replication. These are regions of the genome where the DNA replication machinery is assembled and the DNA is unwound to begin DNA synthesis. In most cases, replication proceeds in both directions from the replication origin.
The main features of replication origins are sequences where specific initiation proteins are bound. A typical replication origin covers about 100-200 base pairs of DNA. Prokaryotes have one origin of replication per chromosome or plasmid but there are usually multiple origins in eukaryotic chromosomes. The human genome contains about 100,000 origins of replication representing about 0.3% of the genome.[25][26][27]
Centromeres

Centromeres are the sites where spindle fibers attach to newly replicated chromosomes in order to segregate them into daughter cells when the cell divides. Each eukaryotic chromosome has a single functional centromere that's seen as a constricted region in a condensed metaphase chromosome. Centromeric DNA consists of a number of repetitive DNA sequences that often take up a significant fraction of the genome because each centromere can be millions of base pairs in length. In humans, for example, the sequences of all 24 centromeres have been determined[29] and they account for about 6% of the genome. However, it's unlikely that all of this noncoding DNA is essential since there is considerable variation in the total amount of centromeric DNA in different individuals.[30] Centromeres are another example of functional noncoding DNA sequences that have been known for almost half a century and it's likely that they are more abundant than coding DNA.
Telomeres
Telomeres are regions of repetitive DNA at the end of a chromosome, which provide protection from chromosomal deterioration during DNA replication. Recent studies have shown that telomeres function to aid in its own stability. Telomeric repeat-containing RNA (TERRA) are transcripts derived from telomeres. TERRA has been shown to maintain telomerase activity and lengthen the ends of chromosomes.[31]
Scaffold attachment regions
Both prokaryotic and eukarotic genomes are organized into large loops of protein-bound DNA. In eukaryotes, the bases of the loops are called scaffold attachment regions (SARs) and they consist of stretches of DNA that bind an RNA/protein complex to stabilize the loop. There are about 100,000 loops in the human genome and each one consists of about 100 bp of DNA. The total amount of DNA devoted to SARs accounts for about 0.3% of the human genome.[32]
Pseudogenes
Pseudogenes are mostly former genes that have become non-functional due to mutation but the term also refers to inactive DNA sequences that are derived from RNAs produced by functional genes (processed pseudogenes). Pseudogenes are only a small fraction of noncoding DNA in prokaryotic genomes because they are eliminated by negative selection. In some eukaryotes, however, pseudogenes can accumulate because selection isn't powerful enough to eliminate them (see Nearly neutral theory of molecular evolution).
The human genome contains about 15,000 pseudogenes derived from protein-coding genes and an unknown number derived from noncoding genes.[33] They may cover a substantial fraction of the genome (~5%) since many of them contain former intron sequences.
Pseudogenes are junk DNA by definition and they evolve at the neutral rate as expected for junk DNA.[34] Some former pseudogenes have secondarily acquired a function and this leads some scientists to speculate that most pseudogenes are not junk because they have a yet-to-be-discovered function.[35]
Repeat sequences, transposons and viral elements
Transposons and retrotransposons are mobile genetic elements. Retrotransposon repeated sequences, which include long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs), account for a large proportion of the genomic sequences in many species. Alu sequences, classified as a short interspersed nuclear element, are the most abundant mobile elements in the human genome. Some examples have been found of SINEs exerting transcriptional control of some protein-encoding genes.[36][37][38]
Endogenous retrovirus sequences are the product of reverse transcription of retrovirus genomes into the genomes of germ cells. Mutation within these retro-transcribed sequences can inactivate the viral genome.[39]
Over 8% of the human genome is made up of (mostly decayed) endogenous retrovirus sequences, as part of the over 42% fraction that is recognizably derived of retrotransposons, while another 3% can be identified to be the remains of DNA transposons. Much of the remaining half of the genome that is currently without an explained origin is expected to have found its origin in transposable elements that were active so long ago (> 200 million years) that random mutations have rendered them unrecognizable.[40] Genome size variation in at least two kinds of plants is mostly the result of retrotransposon sequences.[41][42]
Highly repetitive DNA
Highly repetitive DNA consists of short stretches of DNA that are repeated many times in tandem (one after the other). The repeat segments are usually between 2 bp and 10 bp but longer ones are known. Highly repetitive DNA is rare in prokaryotes but common in eukaryotes, especially those with large genomes. It is sometimes called satellite DNA.
Most of the highly repetitive DNA is found in centromeres and telomeres (see above) and most of it is functional although some might be redundant. The other significant fraction resides in short tandem repeats (STRs; also called microsatellites) consisting of short stretches of a simple repeat such as ATC. There are about 350,000 STRs in the human genome and they are scattered throughout the genome with an average length of about 25 repeats.[43][44]
Variations in the number of STR repeats can cause genetic diseases when they lie within a gene but most of these regions appear to be non-functional junk DNA where the number of repeats can vary considerably from individual to individual. This is why these length differences are used extensively in DNA fingerprinting.
Junk DNA
Junk DNA is DNA that has no biologically relevant function such as pseudogenes and fragments of once active transposons. Bacteria and viral genomes have very little junk DNA[45][46] but some eukaryotic genomes may have a substantial amount of junk DNA.[47] The exact amount of nonfunctional DNA in humans and other species with large genomes has not been determined and there is considerable controversy in the scientific literature.[48][49]
The nonfunctional DNA in bacterial genomes is mostly located in the intergenic fraction of non-coding DNA but in eukaryotic genomes it may also be found within introns. It's important to note that there are many examples of functional DNA elements in non-coding DNA (see above) and there are no scientists who claim that all non-coding DNA is junk.
Genome-wide association studies (GWAS) and non-coding DNA
Genome-wide association studies (GWAS) identify linkages between alleles and observable traits such as phenotypes and diseases. Most of the associations are between single-nucleotide polymorphisms (SNPs) and the trait being examined and most of these SNPs are located in non-functional DNA. The association establishes a linkage that helps map the DNA region responsible for the trait but it doesn't necessarily identify the mutations causing the disease or phenotypic difference.[50][51][52][53][54]
SNPs that are tightly linked to traits are the ones most likely to identify a causal mutation. (The association is referred to as tight linkage disequilibrium.) About 12% of these polymorphisms are found in coding regions; about 40% are located in introns; and most of the rest are found in intergenic regions, including regulatory sequences.[51]
See also
References
- Kirchberger PC, Schmidt ML, and Ochman H (2020). "The ingenuity of bacterial genomes". Annual Review of Microbiology. 74: 815–834. doi:10.1146/annurev-micro-020518-115822. PMID 32692614. S2CID 220699395.
- Piovesan A, Antonaros F, Vitale L, Strippoli P, Pelleri MC, Caracausi M (2019). "Human protein-coding genes and gene feature statistics in 2019". BMC Research Notes. 12 (1): 315. doi:10.1186/s13104-019-4343-8. PMC 6549324. PMID 31164174.
- Omenn GS (2021). "Reflections on the HUPO Human Proteome Project, the Flagship Project of the Human Proteome Organization, at 10 Years". Molecular & Cellular Proteomics. 20: 100062. doi:10.1016/j.mcpro.2021.100062. PMC 8058560. PMID 33640492.
- Thomas CA (1971). "The genetic organization of chromosomes". Annual Review of Genetics. 5: 237–256. doi:10.1146/annurev.ge.05.120171.001321. PMID 16097657.
- Elliott TA, Gregory TR (2015). "What's in a genome? The C-value enigma and the evolution of eukaryotic genome content". Phil. Trans. R. Soc. B. 370 (1678): 20140331. doi:10.1098/rstb.2014.0331. PMC 4571570. PMID 26323762. S2CID 12095046.
- Hahn MW, Wray GA (2002). "The g-value paradox". Evolution and Development. 4 (2): 73–75. doi:10.1046/j.1525-142X.2002.01069.x. PMID 12004964. S2CID 2810069.
- Gregory TR, Hebert PD (April 1999). "The modulation of DNA content: proximate causes and ultimate consequences". Genome Research. 9 (4): 317–324. doi:10.1101/gr.9.4.317. PMID 10207154. S2CID 16791399.
- Aparicio S, Chapman J, Stupka E, Putnam N, Chia JM, Dehal P, Christoffels A, Rash S, Hoon S, Smit A (2002). "Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes". Science. 297 (5585): 1301–1310. Bibcode:2002Sci...297.1301A. doi:10.1126/science.1072104. PMID 12142439. S2CID 10310355.
- Ohno S (1972). "So much "junk" DNA in our genome". Brookhaven Symposia in Biology. 23: 366–370. OCLC 101819442. PMID 5065367.
- Ibarra-Laclette E, Lyons E, Hernández-Guzmán G, Pérez-Torres CA, Carretero-Paulet L, Chang TH, Lan T, Welch AJ, Juárez MJ, Simpson J, et al. (2013). "Architecture and evolution of a minute plant genome". Nature. 498 (7452): 94–98. Bibcode:2013Natur.498...94I. doi:10.1038/nature12132. PMC 4972453. PMID 23665961. S2CID 18219754.
- Lan T, Renner T, Ibarra-Laclette E, Farr KM, Chang TH, Cervantes-Pérez SA, Zheng C, Sankoff D, Tang H, and Purbojati RW (2017). "Long-read sequencing uncovers the adaptive topography of a carnivorous plant genome". Proceedings of the National Academy of Sciences. 114 (22): E4435–E4441. Bibcode:2017PNAS..114E4435L. doi:10.1073/pnas.1702072114. PMC 5465930. PMID 28507139.
- Klein J (19 May 2017). "Genetic Tidying Up Made Humped Bladderworts Into Carnivorous Plants". New York Times. Retrieved May 30, 2022.
- Hsu C, and Stolte D (May 13, 2013). "Carnivorous Plant Throws Out 'Junk' DNA" (Press release). Tucson, AZ, USA: University of Arizona. Retrieved May 29, 2022.
- Kampourakis K (2017). Making sense of genes. Cambridge UK: Cambridge University Press. ISBN 978-1-107-12813-2.
- Cech TR, Steitz JA (2014). "The Noncoding RNA Revolution - Trashing Old Rules to Forge New Ones". Cell. 157 (1): 77–94. doi:10.1016/j.cell.2014.03.008. PMID 24679528. S2CID 14852160.
- Rogozin IB, Makarova KS, Natale DA, Spiridonov AN, Tatusov RL, Wolf YI, et al. (October 2002). "Congruent evolution of different classes of non-coding DNA in prokaryotic genomes". Nucleic Acids Research. 30 (19): 4264–4271. doi:10.1093/nar/gkf549. PMC 140549. PMID 12364605.
- Bielawski JP, Jones C (2016). "Adaptive Molecular Evolution: Detection Methods". Encyclopedia of Evolutionary Biology. pp. 16–25. doi:10.1016/B978-0-12-800049-6.00171-2. ISBN 978-0-12-800426-5.
- Ponting CP, and Haerty W (2022). "Genome-Wide Analysis of Human Long Noncoding RNAs: A Provocative Review". Annual Review of Genomics and Human Genetics. 23: 153–172. doi:10.1146/annurev-genom-112921-123710. hdl:20.500.11820/ede40d70-b99c-42b0-a378-3b9b7b256a1b. PMID 35395170. S2CID 248049706.
- Compe E, Egly JM (2021). "The Long Road to Understanding RNAPII Transcription Initiation and Related Syndromes". Annual Review of Biochemistry. 90: 193–219. doi:10.1146/annurev-biochem-090220-112253. PMID 34153211. S2CID 235595550.
- Visel A, Rubin EM, Pennacchio LA (September 2009). "Genomic views of distant-acting enhancers". Nature. 461 (7261): 199–205. Bibcode:2009Natur.461..199V. doi:10.1038/nature08451. PMC 2923221. PMID 19741700.
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S (2012). "GENCODE: the reference human genome annotation for The ENCODE Project". Genome Research. 22 (9): 1760–1774. doi:10.1101/gr.135350.111. PMC 3431492. PMID 22955987.
- Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson JD (1994). Molecular Biology of the Cell, 3rd edition. London, UK: Garland Publishing Inc.
- Lewin B (2004). Genes VIII. Upper Saddle River, NJ, USA: Pearson/Prentice Hall.
- Moran L, Horton HR, Scrimgeour KG, Perry MD (2012). Principles of Biochemistry Fifth Edition. Upper Saddle River, NJ, USA: Pearson.
- Leonard AC, Méchali M (2013). "DNA replication origins". Cold Spring Harbor Perspectives in Biology. 5 (10): a010116. doi:10.1101/cshperspect.a010116. PMC 3783049. PMID 23838439.
- Urban JM, Foulk MS, Casella C, Gerbi SA (2015). "The hunt for origins of DNA replication in multicellular eukaryotes". F1000Prime Reports. 7: 30. doi:10.12703/P7-30. PMC 4371235. PMID 25926981.
- Prioleau M, MacAlpine DM (2016). "DNA replication origins—where do we begin?". Genes & Development. 30 (15): 1683–1697. doi:10.1101/gad.285114.116. PMC 5002974. PMID 27542827.
- Romiguier J, Roux C (2017). "Analytical Biases Associated with GC-Content in Molecular Evolution". Frontiers in Genetics. 8: 16. doi:10.3389/fgene.2017.00016. PMC 5309256. PMID 28261263.
- Altemose N, Logsdon GA, Bzikadze AV, Sidhwani P, Langley SA, Caldas GV, et al. (2021). "Complete genomic and epigenetic maps of human centromeres". Science. 376 (6588): 56. doi:10.1126/science.abl4178. PMC 9233505. PMID 35357911. S2CID 247853627.
- Miga KH (2019). "Centromeric satellite DNAs: hidden sequence variation in the human population". Genes. 10 (5): 353. doi:10.3390/genes10050352. PMC 6562703. PMID 31072070.
- Cusanelli E, Chartrand P (May 2014). "Telomeric noncoding RNA: telomeric repeat-containing RNA in telomere biology". Wiley Interdisciplinary Reviews. RNA. 5 (3): 407–419. doi:10.1002/wrna.1220. PMID 24523222. S2CID 36918311.
- Mistreli T (2020). "The self-organizing genome: Principles of genome architecture and function". Cell. 183 (1): 28–45. doi:10.1016/j.cell.2020.09.014. PMC 7541718. PMID 32976797.
- "Ensemble Human reference genome GRCh38.p13".
- Xu J, Zhang J (2015). "Are human translated pseudogenes functional?". Molecular Biology and Evolution. 33 (3): 755–760. doi:10.1093/molbev/msv268. PMC 5009996. PMID 26589994.
- Wen YZ, Zheng LL, Qu LH, Ayala FJ, Lun ZR (2012). "Pseudogenes are not pseudo any more". RNA Biology. 9 (1): 27–32. doi:10.4161/rna.9.1.18277. PMID 22258143. S2CID 13161678.
- Ponicsan SL, Kugel JF, Goodrich JA (April 2010). "Genomic gems: SINE RNAs regulate mRNA production". Current Opinion in Genetics & Development. 20 (2): 149–155. doi:10.1016/j.gde.2010.01.004. PMC 2859989. PMID 20176473.
- Häsler J, Samuelsson T, Strub K (July 2007). "Useful 'junk': Alu RNAs in the human transcriptome". Cellular and Molecular Life Sciences (Submitted manuscript). 64 (14): 1793–1800. doi:10.1007/s00018-007-7084-0. PMID 17514354. S2CID 5938630.
- Walters RD, Kugel JF, Goodrich JA (August 2009). "InvAluable junk: the cellular impact and function of Alu and B2 RNAs". IUBMB Life. 61 (8): 831–837. doi:10.1002/iub.227. PMC 4049031. PMID 19621349.
- Nelson PN, Hooley P, Roden D, Davari Ejtehadi H, Rylance P, Warren P, et al. (October 2004). "Human endogenous retroviruses: transposable elements with potential?". Clinical and Experimental Immunology. 138 (1): 1–9. doi:10.1111/j.1365-2249.2004.02592.x. PMC 1809191. PMID 15373898.
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. (February 2001). "Initial sequencing and analysis of the human genome". Nature. 409 (6822): 860–921. Bibcode:2001Natur.409..860L. doi:10.1038/35057062. PMID 11237011.
- Piegu B, Guyot R, Picault N, Roulin A, Sanyal A, Saniyal A, et al. (October 2006). "Doubling genome size without polyploidization: dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice". Genome Research. 16 (10): 1262–1269. doi:10.1101/gr.5290206. PMC 1581435. PMID 16963705.
- Hawkins JS, Kim H, Nason JD, Wing RA, Wendel JF (October 2006). "Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium". Genome Research. 16 (10): 1252–1261. doi:10.1101/gr.5282906. PMC 1581434. PMID 16954538.
- Gymrek M, Willems T, Guilmatre A, Zeng H, Markus B, Georgiev S, Daly MJ, Price AL, Pritchard JK, Sharp AJ, Erlich Y (2016). "Abundant contribution of short tandem repeats to gene expression variation in humans". Nature Genetics. 48 (1): 22–29. doi:10.1038/ng.3461. PMC 4909355. PMID 26642241.
- Kronenberg ZN, Fiddes IT, Gordon D, Murali S, Cantsilieris S, Meyerson OS, Underwood JG, Nelson BJ, Chaisson MJ, Dougherty ML (2018). "High-resolution comparative analysis of great ape genomes". Science. 360 (6393): 1085. doi:10.1126/science.aar6343. PMC 6178954. PMID 29880660.
- Gil R, and Latorre A (2012). "Factors behind junk DNA in bacteria". Genes. 3 (4): 634–650. doi:10.3390/genes3040634. PMC 3899985. PMID 24705080.
- Brandes, Nadav; Linial, Michal (2016). "Gene overlapping and size constraints in the viral world". Biology Direct. 11 (1): 26. doi:10.1186/s13062-016-0128-3. ISSN 1745-6150. PMC 4875738. PMID 27209091.
- Palazzo AF, Gregory TR (May 2014). "The case for junk DNA". PLOS Genetics. 10 (5): e1004351. doi:10.1371/journal.pgen.1004351. PMC 4014423. PMID 24809441.
- Morange, Michel (2014). "Genome as a Multipurpose Structure Built by Evolution" (PDF). Perspectives in Biology and Medicine. 57 (1): 162–171. doi:10.1353/pbm.2014.0008. PMID 25345709. S2CID 27613442.
- Haerty W, and Ponting CP (2014). "No Gene in the Genome Makes Sense Except in the Light of Evolution". Annual Review of Genomics and Human Genetics. 25: 71–92. doi:10.1146/annurev-genom-090413-025621. PMID 24773316.
- Korte A, Farlwo A (2013). "The advantages and limitations of trait analysis with GWAS: a review". Plant Methods. 9: 29. doi:10.1186/1746-4811-9-29. PMC 3750305. PMID 23876160. S2CID 206976469.
- Manolio TA (July 2010). "Genomewide association studies and assessment of the risk of disease". The New England Journal of Medicine. 363 (2): 166–76. doi:10.1056/NEJMra0905980. PMID 20647212.
- Visscher PV, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, Yang J (2017). "10 Years of GWAS Discovery: Biology, Function, and Translation". American Journal of Human Genetics. 101 (1): 5–22. doi:10.1016/j.ajhg.2017.06.005. PMC 5501872. PMID 28686856.
- Gallagher MD, Chen-Plotkin, AS (2018). "The Post-GWAS Era: From Association to Function". American Journal of Human Genetics. 102 (5): 717–730. doi:10.1016/j.ajhg.2018.04.002. PMC 5986732. PMID 29727686.
- Marigorta UM, Rodríguez JA, Gibson G, Navarro A (2018). "Replicability and Prediction: Lessons and Challenges from GWAS". Trends in Genetics. 34 (7): 504–517. doi:10.1016/j.tig.2018.03.005. PMC 6003860. PMID 29716745.
Further reading
- Bennett MD, Leitch IJ (2005). "Genome size evolution in plants". In Gregory RT (ed.). The Evolution of the Genome. San Diego: Elsevier. pp. 89–162. ISBN 978-0-08-047052-8.
- Gregory TR (2005). "Genome Size Evolution in Animals". The Evolution of the Genome. pp. 3–87. doi:10.1016/B978-012301463-4/50003-6. ISBN 978-0-12-301463-4.
- Shabalina SA, Spiridonov NA (2004). "The mammalian transcriptome and the function of non-coding DNA sequences". Genome Biology. 5 (4): 105. doi:10.1186/gb-2004-5-4-105. PMC 395773. PMID 15059247.
- Castillo-Davis CI (October 2005). "The evolution of noncoding DNA: how much junk, how much func?". Trends in Genetics. 21 (10): 533–536. doi:10.1016/j.tig.2005.08.001. PMID 16098630.