Nucleic acid metabolism
Nucleic acid metabolism is a collective term that refers to the variety of chemical reactions by which nucleic acids (DNA and/or RNA) are either synthesized or degraded. Nucleic acids are polymers (so-called "biopolymers") made up of a variety of monomers called nucleotides. Nucleotide synthesis is an anabolic mechanism generally involving the chemical reaction of phosphate, pentose sugar, and a nitrogenous base. Degradation of nucleic acids is a catabolic reaction and the resulting parts of the nucleotides or nucleobases can be salvaged to recreate new nucleotides. Both synthesis and degradation reactions require multiple enzymes to facilitate the event. Defects or deficiencies in these enzymes can lead to a variety of diseases.[1]
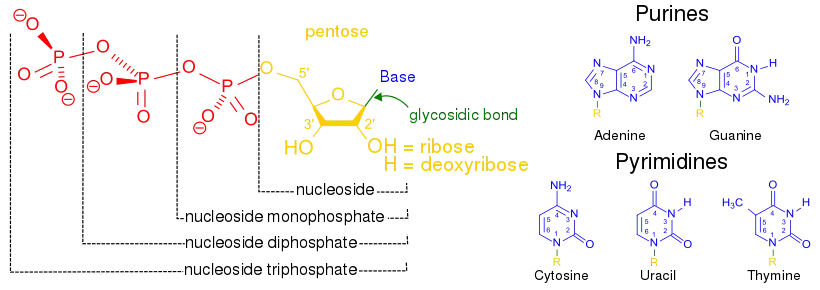
Synthesis of nucleotides
Nucleotides are the monomers which polymerize into nucleic acids. All nucleotides contain a sugar, a phosphate, and a nitrogenous base. The bases found in nucleic acids are either purines or pyrimidines. In the more complex multicellular animals, they are both primarily produced in the liver but the two different groups are synthesized in different ways. However, all nucleotide synthesis requires the use of phosphoribosyl pyrophosphate (PRPP) which donates the ribose and phosphate necessary to create a nucleotide.
Purine synthesis
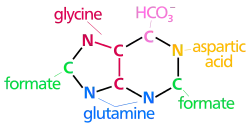
Adenine and guanine are the two nucleotides classified as purines. In purine synthesis, PRPP is turned into inosine monophosphate, or IMP. Production of IMP from PRPP requires glutamine, glycine, aspartate, and 6 ATP, among other things.[1] IMP is then converted to AMP (adenosine monophosphate) using GTP and aspartate, which is converted into fumarate. While IMP can be directly converted to AMP, synthesis of GMP (guanosine monophosphate) requires an intermediate step, in which NAD+ is used to form the intermediate xanthosine monophosphate, or XMP. XMP is then converted into GMP by using the hydrolysis of 1 ATP and the conversion of glutamine to glutamate.[1] AMP and GMP can then be converted into ATP and GTP, respectively, by kinases that add additional phosphates.
ATP stimulates production of GTP, while GTP stimulates production of ATP. This cross regulation keeps the relative amounts of ATP and GTP the same. Excess of either nucleotide could increase the likelihood of DNA mutations, where the wrong purine nucleotide is inserted.[1]
Lesch–Nyhan syndrome is caused by a deficiency in hypoxanthine-guanine phosphoribosyltransferase or HGPRT, the enzyme that catalyzes the reversible reaction of producing guanine from GMP. This is a sex-linked congenital defect that causes overproduction of uric acid along with mental retardation, spasticity, and an urge to self-mutilate.[1][2][3]
Pyrimidine synthesis
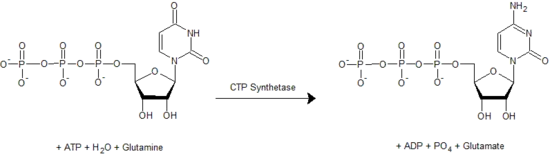
Pyrimidine nucleosides include cytidine, uridine, and thymidine.[4] The synthesis of any pyrimidine nucleotide begins with the formation of uridine. This reaction requires aspartate, glutamine, bicarbonate, and 2 ATP molecules (to provide energy), as well as PRPP which provides ribose-monophosphate. Unlike in purine synthesis, the sugar/phosphate group from PRPP is not added to the nitrogenous base until towards the end of the process. After synthesizing uridine-monophosphate, it can react with 2 ATP to form uridine-triphosphate or UTP. UTP can be converted to CTP (cytidine-triphosphate) in a reaction catalyzed by CTP synthetase. Thymidine synthesis first requires reduction of the uridine to deoxyuridine (see next section), before the base can be methylated to produce thymidine. [1][5]
ATP, a purine nucleotide, is an activator of pyrimidine synthesis, while CTP, a pyrimidine nucleotide, is an inhibitor of pyrimidine synthesis. This regulation helps to keep the purine/pyrimidine amounts similar, which is beneficial because equal amounts of purines and pyrimidines are required for DNA synthesis.[1][6]
Deficiencies of enzymes involved in pyrimidine synthesis can lead to the genetic disease Orotic aciduria which causes excessive excretion of orotic acid in the urine.[1][7]
Converting nucleotides to deoxynucleotides
Nucleotides are initially made with ribose as the sugar component, which is a feature of RNA. DNA, however, requires deoxyribose, which is missing the 2'-hydroxyl (-OH group) on the ribose. The reaction to remove this -OH is catalyzed by ribonucleotide reductase. This enzyme converts NDPs (nucleoside-diphosphate) to dNDPs (deoxynucleoside-diphosphate). The nucleotides must be in the diphosphate form for the reaction to occur.[1]
In order to synthesize thymidine, a component of DNA which only exists in the deoxy form, uridine is converted to deoxyuridine (by ribonucleotide reductase), and then is methylated by thymidylate synthase to create thymidine.[1]
Degradation of nucleic acids
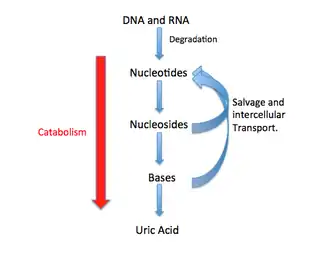
The breakdown of DNA and RNA is occurring continuously in the cell. Purine and pyrimidine nucleosides can either be degraded to waste products and excreted or can be salvaged as nucleotide components.[5]
Pyrimidine catabolism
Cytosine and uracil are converted into beta-alanine and later to malonyl-CoA which is needed for fatty acid synthesis, among other things. Thymine, on the other hand, is converted into β-aminoisobutyric acid which is then used to form methylmalonyl-CoA. The leftover carbon skeletons such as acetyl-CoA and Succinyl-CoA can then be oxidized by the citric acid cycle. Pyrimidine degradation ultimately ends in the formation of ammonium, water, and carbon dioxide. The ammonium can then enter the urea cycle which occurs in the cytosol and the mitochondria of cells.[5]
Pyrimidine bases can also be salvaged. For example, the uracil base can be combined with ribose-1-phosphate to create uridine monophosphate or UMP. A similar reaction can also be done with thymine and deoxyribose-1-phosphate.[8]
Deficiencies in enzymes involved in pyrimidine catabolism can lead to diseases such as Dihydropyrimidine dehydrogenase deficiency which has negative neurological effects.[9]
Purine catabolism
Purine degradation takes place mainly in the liver of humans and requires an assortment of enzymes to degrade purines to uric acid. First, the nucleotide will lose its phosphate through 5'-nucleotidase. The nucleoside, adenosine, is then deaminated and hydrolyzed to form hypoxanthine via adenosine deaminase and nucleosidase respectively. Hypoxanthine is then oxidized to form xanthine and then uric acid through the action of xanthine oxidase. The other purine nucleoside, guanosine, is cleaved to form guanine. Guanine is then deaminated via guanine deaminase to form xanthine which is then converted to uric acid. Oxygen is the final electron acceptor in the degradation of both purines. Uric acid is then excreted from the body in different forms depending on the animal.[5]
Free purine and pyrimidine bases that are released into the cell are typically transported intercellularly across membranes and salvaged to create more nucleotides via nucleotide salvage. For example, adenine + PRPP --> AMP + PPi. This reaction requires the enzyme adenine phosphoribosyltransferase. Free guanine is salvaged in the same way except it requires hypoxanthine-guanine phosphoribosyltransferase.
Defects in purine catabolism can result in a variety of diseases including gout, which stems from an accumulation of uric acid crystals in various joints, and adenosine deaminase deficiency, which causes immunodeficiency.[10][11][12]
Interconversion of nucleotides
Once the nucleotides are synthesized they can exchange phosphates among one another in order to create mono-, di-, and tri-phosphate molecules. The conversion of a nucleoside-diphosphate (NDP) to a nucleoside-triphosphate (NTP) is catalyzed by nucleoside diphosphate kinase, which uses ATP as the phosphate donor. Similarly, nucleoside-monophosphate kinase carries out the phosphorylation of nucleoside-monophosphates. Adenylate kinase is a specific nucleoside-monophosphate kinase that functions only on adenosine-monophosphate.[1][8]
References
- Voet, Donald; Voet, Judith; Pratt, Charlotte (2008). Fundamentals of the biochemistry : life at the molecular level (3rd ed.). Hoboken, NJ: Wiley. ISBN 9780470129302.
- Nyhan, WL (1973). "The Lesch-Nyhan syndrome". Annual Review of Medicine. 24: 41–60. doi:10.1146/annurev.me.24.020173.000353. PMID 4575865.
- "Lesch-Nyhan". Lesch-Nyhan.org. Retrieved 31 October 2014.
- Alqahtani, Saad Saeed; Koltai, Tomas; Ibrahim, Muntaser E.; Bashir, Adil H. H.; Alhoufie, Sari T. S.; Ahmed, Samrein B. M.; Molfetta, Daria Di; Carvalho, Tiago M. A.; Cardone, Rosa Angela; Reshkin, Stephan Joel; Hifny, Abdelhameed; Ahmed, Mohamed E.; Alfarouk, Khalid Omer (6 July 2022). "Role of pH in Regulating Cancer Pyrimidine Synthesis". Journal of Xenobiotics. 12 (3): 158–180. doi:10.3390/jox12030014. PMC 9326563. PMID 35893264.
- Nelson, David L.; Cox, Michael M.; Lehninger, Albert L. (2008). Lehninger's Principles of Biochemistry (5 ed.). Macmillan. ISBN 978-0716771081.
- "Nucleotide Metabolism II". Oregon State. Archived from the original on 11 February 2017. Retrieved 20 October 2014.
- Bailey, CJ (2009). "Orotic aciduria and uridine monophosphate synthase: a reappraisal". Journal of Inherited Metabolic Disease. 32: S227-33. doi:10.1007/s10545-009-1176-y. PMID 19562503. S2CID 13215215.
- "Nucleotide Metabolism". The Medical Biochemistry Page. Retrieved 20 October 2014.
- "Dihydropyrimidine dehydrogenase deficiency". Genetics Home Reference. Retrieved 31 October 2014.
- "Nucleotides: Their Synthesis and Degradation". Molecular Biochemistry II. Retrieved 20 October 2014.
- Kelley, RE; Andersson, HC (2014). "Disorders of purines and pyrimidines". Handbook of Clinical Neurology. 120: 827–38. doi:10.1016/B978-0-7020-4087-0.00055-3. ISBN 9780702040870. PMID 24365355.
- "Adenosine deaminase (ADA) deficiency". Learn.Genetics. Archived from the original on 3 November 2014. Retrieved 31 October 2014.
External links
- Nucleic Acids Book (free online book on the chemistry and biology of nucleic acids)
- Interactive overview of nucleic acid metabolism.
Metabolism map | ||
|---|---|---|
Single lines: pathways common to most lifeforms. Double lines: pathways not in humans (occurs in e.g. plants, fungi, prokaryotes). | ||
.svg.png.webp)