Purine nucleotide cycle
The Purine Nucleotide Cycle is a metabolic pathway in protein metabolism requiring the amino acids aspartate and glutamate. The cycle is used to regulate the levels of adenine nucleotides, in which ammonia and fumarate are generated.[2] AMP coverts into IMP and the byproduct ammonia. IMP converts to S-AMP (adenylosuccinate), which then coverts to AMP and the byproduct fumarate. The fumarate goes on to produce ATP (energy) via oxidative phosphorylation as it enters the Krebs cycle and then the electron transport chain. Lowenstein first described this pathway and outlined its importance in processes including amino acid catabolism and regulation of flux through glycolysis and the Krebs cycle.[2][3][4]
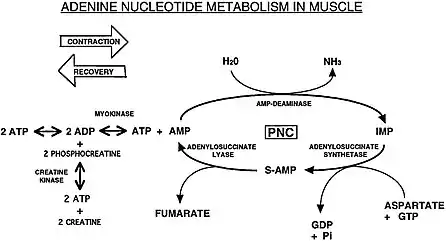
AMP is produced after strenuous muscle contraction when the ATP reservoir is low (ADP > ATP) by the adenylate kinase (myokinase) reaction.[5][6] AMP is also produced from adenine and adenosine directly; however, AMP can be produced through less direct metabolic pathways, such as de novo synthesis of IMP or through salvage pathways of guanine (a purine) and any of the purine nucleotides and nucleosides. IMP is synthesized de novo from glucose through the pentose phosphate pathway which produces ribose 5-P, which then converts to PRPP that with the amino acids glycine, glutamine, and aspartate (see Purine metabolism) can be further converted into IMP.[7]
Reactions
The cycle comprises three enzyme-catalysed reactions. The first stage is the deamination of the purine nucleotide adenosine monophosphate (AMP) to form inosine monophosphate (IMP), catalysed by the enzyme AMP deaminase:
- AMP + H2O + H+ → IMP + NH3
The second stage is the formation of adenylosuccinate from IMP and the amino acid aspartate, which is coupled to the energetically favourable hydrolysis of GTP, and catalysed by the enzyme adenylosuccinate synthetase:
Finally, adenylosuccinate is cleaved by the enzyme adenylosuccinate lyase to release fumarate and regenerate the starting material of AMP:
- Adenylosuccinate → AMP + Fumarate
A recent study showed that activation of HIF-1α allows cardiomyocytes to sustain mitochondrial membrane potential during anoxic stress by utilizing fumarate produced by adenylosuccinate lyase as an alternate terminal electron acceptor in place of oxygen. This mechanism should help provide protection in the ischemic heart.[8]
Occurrence
The purine nucleotide cycle occurs in the cytosol (a gel-like substance) of the sarcoplasm of skeletal muscle, and in the myocyte's cytosolic compartment of the cytoplasm of cardiac and smooth muscle. The cycle occurs when ATP reservoirs run low (ADP > ATP), such as strenuous exercise, fasting or starvation.[5][9]
Proteins catabolize into amino acids, and amino acids are precursors for purines, nucleotides and nucleosides which are used in the purine nucleotide cycle.[7] The amino acid glutamate is used to neutralize the ammonia produced when AMP is converted into IMP. Another amino acid, aspartate, is used along with IMP to produce S-AMP in the cycle. Skeletal muscle contains amino acids for use in catabolism, known as the free amino acid pool; however, inadequate carbohydrate supply and/or strenuous exercise requires protein catabolism to sustain the free amino acids.[9]
When the phosphagen system (ATP-PCr) has been depleted of phosphocreatine (creatine phosphate), the purine nucleotide cycle also helps to sustain the myokinase reaction by reducing accumulation of AMP produced after muscle contraction in the below reaction.[6]
During muscle contraction:
- H2O + ATP → H+ + ADP + Pi (Mg2+ assisted, utilization of ATP for muscle contraction by ATPase)
- H+ + ADP + CP → ATP + Creatine (Mg2+ assisted, catalyzed by creatine kinase, ATP is used again in the above reaction for continued muscle contraction)
- 2 ADP → ATP + AMP (catalyzed by adenylate kinase/myokinase when CP is depleted, ATP is again used for muscle contraction)
Muscle at rest:
- ATP + Creatine → H+ + ADP + CP (Mg2+ assisted, catalyzed by creatine kinase)
- ADP + Pi → ATP (during anaerobic glycolysis and oxidative phosphorylation)
AMP can dephosphorylate to adenosine and diffuse out of the cell; the purine nucleotide cycle may therefore also reduce the loss of adenosine from the cell since nucleosides permeate cell membranes, whereas nucleotides do not.[6]
Consequences
Aspartate and glutamate synthesis

Fumarate, produced from the purine nucleotide cycle, is an intermediate of TCA cycle and enters the mitochondria by converting into malate and utilizing the malate shuttle where it is converted into oxaloacetic acid (OAA). During exercise, OAA either enters into TCA cycle or converts into aspartate in the mitochondria.[10]
As the purine nucleotide cycle produces ammonia (see below in ammonia synthesis), skeletal muscle needs to synthesize glutamate in a way that does not further increase ammonia, and as such the use of glutaminase to produce glutamate from glutamine would not be ideal. Also, plasma glutamine (released from the kidneys) requires active transport into the muscle cell (consuming ATP).[11] Consequently, during exercise when the ATP reservoir is low (ADP>ATP), glutamate is produced from branch-chained amino acids (BCAAs) and α-ketoglutarate, as well as from alanine and α-ketoglutarate.[12] Glutamate is then used to produce aspartate. The aspartate enters the purine nucleotide cycle, where it is used to convert IMP into S-AMP.[10][13]
- BCAAs + α-Ketoglutarate ⇌ Glutamate + Branch-chain keto acids (BCKAs) (catalyzed by Branched-chain aminotransferases (BCAT))
- Alanine + α-Ketoglutarate ⇌ Pyruvate + Glutamate (catalyzed by alanine transaminase)
- Oxaloacetic acid + Glutamate ⇌ α-Ketoglutarate + Aspartate (catalyzed by aspartate aminotransferase)
When skeletal muscle is at rest (ADP<ATP), the aspartate is no longer needed for the purine nucleotide cycle and can therefore be used with α-ketoglutarate to produce glutamate and oxaloacetic acid (the above reaction reversed).
α-Ketoglutarate + Aspartate ⇌ Oxaloacetic acid + Glutamate (catalyzed by aspartate aminotransferase)
Ammonia and glutamine synthesis
During exercise when the ATP reservoir is low (ADP>ATP), the purine nucleotide cycle produces ammonia (NH3) when it converts AMP into IMP. (With the exception of AMP deaminase deficiency, where ammonia is produced during exercise when adenosine, from AMP, is converted into inosine). During rest (ADP<ATP), ammonia is produced from the conversion of adenosine into inosine by adenosine deaminase.
- AMP + H2O + H+ → IMP + NH3 (catalyzed by AMP deaminase in skeletal muscle)
- Adenosine + H2O → Inosine + NH3 (catalyzed by adenosine deaminase in skeletal muscle, blood, liver)
Ammonia is toxic, disrupts cell function, and permeates cell membranes. Ammonia becomes ammonium (NH4+) depending on the pH of the cell or plasma. Ammonium is relatively non-toxic and does not readily permeate cell membranes.[14]
NH3 + H+ ⇌ NH4+
Ammonia (NH3) diffuses into the blood, circulating to the liver to be neutralized by the urea cycle. (N.b. urea is not the same as uric acid, though both are end products of the purine nucleotide cycle, from ammonia and nucleotides respectively.) When the skeletal muscles are at rest (ADP<ATP), ammonia (NH3) combines with glutamate to produce glutamine, which is an energy-consuming step, and the glutamine enters the blood.[15][11]
Glutamate + NH3 + ATP → Glutamine + ADP + Pi (catalyzed by glutamine synthetase in resting skeletal muscle)
Excess glutamine is used by proximal tubule in the kidneys for ammoniagenesis, which may counteract any metabolic acidosis from anaerobic skeletal muscle activity.[15] In kidneys, glutamine is deaminated twice to form glutamate and then α-ketoglutarate. These NH3 molecules neutralise the organic acids (lactic acid and ketone bodies) produced in the muscles.
Glutamine + H2O → Glutamate + NH4+ (catalyzed by glutaminase in the kidneys)
Pathology
Some metabolic myopathies involve the under- or over-utilization of the purine nucleotide cycle. Metabolic myopathies cause a low ATP reservoir in muscle cells (ADP > ATP), resulting in exercise-induced excessive AMP buildup in muscle, and subsequent exercise-induced hyperuricemia (myogenic hyperuricemia) through conversion of excessive AMP into uric acid by way of either AMP → adenosine or AMP → IMP.
During strenuous exercise, AMP is created through the use of the adenylate kinase (myokinase) reaction after the phosphagen system has been depleted of creatine phosphate and not enough ATP is being produced yet by other pathways (see above reaction in 'Occurrence' section). In those affected by metabolic myopathies, exercise that normally wouldn't be considered strenuous for healthy people, is however strenuous for them due to their low ATP reservoir in muscle cells. This results in regular use of the myokinase reaction for normal, everyday activities.
Besides the myokinase reaction, a high ATP consumption and low ATP reservoir also increases protein catabolism and salvage of IMP, which results in increased AMP and IMP. These two nucleotides can then enter the purine nucleotide cycle to produce fumarate which will then produce ATP by oxidative phosphorylation. If the purine nucleotide cycle is blocked (such as AMP deaminase deficiency) or if exercise is stopped and increased fumarate production is no longer needed, then the excess nucleotides will be converted into uric acid.
AMP deaminase deficiency (MADD)
AMP deaminase deficiency (formally known as myoadenylate deaminase deficiency or MADD) is a metabolic myopathy which results in excessive AMP buildup brought on by exercise. AMP deaminase is needed to convert AMP into IMP in the purine nucleotide cycle. Without this enzyme, the excessive AMP buildup is initially due to the adenylate kinase (myokinase) reaction which occurs after a muscle contraction.[16] However, AMP is also used to allosterically regulate the enzyme myophosphorylase (see Glycogen phosphorylase § Regulation), so the initial buildup of AMP triggers the enzyme myophosphorylase to release muscle glycogen into glucose-1-P (glycogen→glucose-1-P),[17] which eventually depletes the muscle glycogen, which in turn triggers protein metabolism, which then produces even more AMP. In AMP deaminase deficiency, excess adenosine is converted into uric acid in the following reaction:
- AMP → Adenosine → Inosine → Hypoxanthine → Xanthine → Uric Acid
Glycogenoses (GSDs)
Myogenic hyperuricemia, as a result of the purine nucleotide cycle running when ATP reservoirs in muscle cells are low (ADP > ATP), is a common pathophysiologic feature of glycogenoses such as GSD-III, GSD-V and GSD-VII, as they are metabolic myopathies which impair the ability of ATP (energy) production within muscle cells. In these metabolic myopathies, myogenic hyperuricemia is exercise-induced; inosine, hypoxanthine and uric acid increase in plasma after exercise and decrease over hours with rest.[18] Excess AMP (adenosine monophosphate) is converted into uric acid.[18]
- AMP → IMP → Inosine → Hypoxanthine → Xanthine → Uric acid
Hyperammonemia is also seen post-exercise in McArdle disease (GSD-V) and phosphoglucomutase deficiency (PGM1-CDG, formerly GSD-XIV), due to the purine nucleotide cycle running when the ATP reservoir is low due to the glycolytic block.[19][20][21][22][23][24]
AMP + H2O + H+ → IMP + NH3
See also
References
- Lewis, Amy & Guicherit, Oivin & Datta, Surjit & Hanten, Gerri & Kellems, Rodney. (1996). Structure and Expression of the Murine Muscle Adenylosuccinate Synthetase Gene. The Journal of biological chemistry. 271. 22647-56. 10.1074/jbc.271.37.22647.
- Lowenstein J.M. (1972). "Ammonia Production in Muscle and Other Tissues: the Purine Nucleotide Cycle". Physiological Reviews. 52 (2): 382–414. doi:10.1152/physrev.1972.52.2.382. PMID 4260884.
- Salway, J. G. (2004). Metabolism at a glance (3rd ed.). Malden, Mass.: Blackwell Pub. ISBN 1-4051-0716-2. OCLC 53178315.
- Voet, Donald (2011). Biochemistry. Voet, Judith G. (4th ed.). Hoboken, NJ: John Wiley & Sons. ISBN 978-0-470-57095-1. OCLC 690489261.
- Valberg, Stephanie J. (2008-01-01), Kaneko, J. Jerry; Harvey, John W.; Bruss, Michael L. (eds.), "Chapter 15 - Skeletal Muscle Function", Clinical Biochemistry of Domestic Animals (Sixth Edition), San Diego: Academic Press, pp. 459–484, ISBN 978-0-12-370491-7, retrieved 2023-10-10
- N.V. Bhagavan, Chung-Eun Ha, in Essentials of Medical Biochemistry (Second Edition), 2015. Chapter 19 - Contractile Systems: Reaction and the Purine Nucleotide Cycle
- "Purine Synthesis : Synthesis of Purine RiboNucleotides". 2022-04-29. Retrieved 2022-12-14.
- Sridharan et al. (AJP Cell Physiology, 2008, 295:C29-C37)
- Baker JS, McCormick MC, Robergs RA. Interaction among Skeletal Muscle Metabolic Energy Systems during Intense Exercise. J Nutr Metab. 2010;2010:905612. doi: 10.1155/2010/905612. Epub 2010 Dec 6. PMID 21188163; PMCID: PMC3005844.
- Arinze, Ifeanyi J. (May 2005). "Facilitating understanding of the purine nucleotide cycle and the one-carbon pool: Part I: The purine nucleotide cycle". Biochemistry and Molecular Biology Education. 33 (3): 165–168. doi:10.1002/bmb.2005.494033032469. PMID 21638570. S2CID 35570434.
- Cruzat, Vinicius; Macedo Rogero, Marcelo; Noel Keane, Kevin; Curi, Rui; Newsholme, Philip (2018-10-23). "Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation". Nutrients. 10 (11): 1564. doi:10.3390/nu10111564. ISSN 2072-6643. PMC 6266414. PMID 30360490.
- Wagenmakers, A. J. (1998). "Muscle amino acid metabolism at rest and during exercise: role in human physiology and metabolism". Exercise and Sport Sciences Reviews. 26: 287–314. doi:10.1249/00003677-199800260-00013. ISSN 0091-6331. PMID 9696993. S2CID 21127358.
- Mann, Gagandeep; Mora, Stephen; Madu, Glory; Adegoke, Olasunkanmi A. J. (2021). "Branched-chain Amino Acids: Catabolism in Skeletal Muscle and Implications for Muscle and Whole-body Metabolism". Frontiers in Physiology. 12: 702826. doi:10.3389/fphys.2021.702826. ISSN 1664-042X. PMC 8329528. PMID 34354601.
- Wilson, David A., ed. (2012-01-01), "Ammonia", Clinical Veterinary Advisor, Saint Louis: W.B. Saunders, p. 905, doi:10.1016/b978-1-4160-9979-6.00355-x, ISBN 978-1-4160-9979-6, retrieved 2023-03-22
- Taneja, Vikas; Jasuja, Haneesh (November 2019). "Severe hyperammonemia from intense skeletal muscle activity: A rare case report and literature review". Medicine. 98 (47): e17981. doi:10.1097/MD.0000000000017981. ISSN 0025-7974. PMC 6882587. PMID 31764807. S2CID 208276909.
- Sabina, Richard L.; Swain, Judith L.; Patten, Bernard M.; Ashizawa, Tetsuo; O'Brien, William E.; Holmes, Edward W. (1980-12-01). "Disruption of the Purine Nucleotide Cycle". Journal of Clinical Investigation. 66 (6): 1419–1423. doi:10.1172/JCI109995. ISSN 0021-9738. PMC 371628. PMID 7440723.
- Meyer, Francois; Heilmeyer, Ludwig M.G.; Haschke, Richard H.; Fischer, Edmond H. (1970-12-25). "Control of Phosphorylase Activity in a Muscle Glycogen Particle". Journal of Biological Chemistry. 245 (24): 6642–6648. doi:10.1016/s0021-9258(18)62582-7. ISSN 0021-9258. S2CID 33852514.
- Mineo, Ikuo; Kono, Norio; Hara, Naoko; Shimizu, Takao; Yamada, Yuya; Kawachi, Masanori; Kiyokawa, Hiroaki; Wang, Yan Lin; Tarui, Seiichiro (1987-07-09). "Myogenic Hyperuricemia". New England Journal of Medicine. 317 (2): 75–80. doi:10.1056/NEJM198707093170203. ISSN 0028-4793. PMID 3473284.
- Altassan, Ruqaiah; Radenkovic, Silvia; Edmondson, Andrew C.; Barone, Rita; Brasil, Sandra; Cechova, Anna; Coman, David; Donoghue, Sarah; Falkenstein, Kristina; Ferreira, Vanessa; Ferreira, Carlos; Fiumara, Agata; Francisco, Rita; Freeze, Hudson; Grunewald, Stephanie (January 2021). "International consensus guidelines for phosphoglucomutase 1 deficiency (PGM1-CDG): Diagnosis, follow-up, and management". Journal of Inherited Metabolic Disease. 44 (1): 148–163. doi:10.1002/jimd.12286. ISSN 0141-8955. PMC 7855268. PMID 32681750.
- Stojkovic, Tanya; Vissing, John; Petit, François; Piraud, Monique; Orngreen, Mette C.; Andersen, Grete; Claeys, Kristl G.; Wary, Claire; Hogrel, Jean-Yves; Laforêt, Pascal (2009-07-23). "Muscle Glycogenosis Due to Phosphoglucomutase 1 Deficiency". New England Journal of Medicine. 361 (4): 425–427. doi:10.1056/NEJMc0901158. ISSN 0028-4793. PMID 19625727.
- Jean-Yves Hogrel, Jorien B E Janssen, Isabelle Ledoux, Gwenn Ollivier, Anthony Béhin, et al.. The diagnostic value of hyperammonaemia induced by the non-ischaemic forearm exercise test. Journal of Clinical Pathology, 2017, 70 (10), pp.896 - 898. 10.1136/jclinpath-2017-204324 . hal-01618833
- Rumpf, K. W.; Wagner, H.; Kaiser, H.; Meinck, H. M.; Goebel, H. H.; Scheler, F. (1981-12-01). "Increased ammonia production during forearm ischemic work test in McArdle's disease". Klinische Wochenschrift. 59 (23): 1319–1320. doi:10.1007/BF01711182. ISSN 0023-2173. PMID 6947119. S2CID 25192276.
- Vissing, J.; MacLean, D. A.; Vissing, S. F.; Sander, M.; Saltin, B.; Haller, R. G. (2001-12-01). "The exercise metaboreflex is maintained in the absence of muscle acidosis: insights from muscle microdialysis in humans with McArdle's disease". The Journal of Physiology. 537 (Pt 2): 641–649. doi:10.1111/j.1469-7793.2001.00641.x. ISSN 0022-3751. PMC 2278977. PMID 11731594.
- Wagenmakers, A. J.; Coakley, J. H.; Edwards, R. H. (May 1990). "Metabolism of branched-chain amino acids and ammonia during exercise: clues from McArdle's disease". International Journal of Sports Medicine. 11 Suppl 2: S101–113. doi:10.1055/s-2007-1024861. ISSN 0172-4622. PMID 2193889.