Organic anion transporter 1
The organic anion transporter 1 (OAT1) also known as solute carrier family 22 member 6 (SLC22A6) is a protein that in humans is encoded by the SLC22A6 gene.[2][3][4] It is a member of the organic anion transporter (OAT) family of proteins. OAT1 is a transmembrane protein that is expressed in the brain, the placenta, the eyes, smooth muscles, and the basolateral membrane of proximal tubular cells of the kidneys. It plays a central role in renal organic anion transport. Along with OAT3, OAT1 mediates the uptake of a wide range of relatively small and hydrophilic organic anions from plasma into the cytoplasm of the proximal tubular cells of the kidneys. From there, these substrates are transported into the lumen of the nephrons of the kidneys for excretion. OAT1 homologs have been identified in rats, mice, rabbits, pigs, flounders, and nematodes.[5]
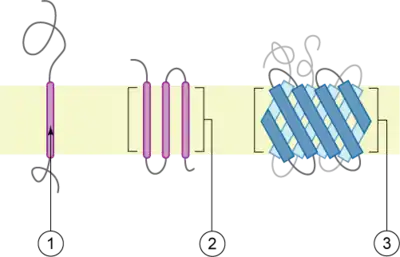
Schematic representation of transmembrane proteins:
1. a membrane protein with one transmembrane domain
2. a membrane protein with three transmembrane domains
3. OAT1 is believed to have twelve transmembrane domains.[1]
The membrane is represented in light brown.
Function
| SLC22A6 | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Aliases | SLC22A6, HOAT1, OAT1, PAHT, ROAT1, Organic anion transporter 1, solute carrier family 22 member 6 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| External IDs | OMIM: 607582 MGI: 892001 HomoloGene: 16813 GeneCards: SLC22A6 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||


OAT1 functions as organic anion exchanger. When the uptake of one molecule of an organic anion is transported into a cell by an OAT1 exchanger, one molecule of an endogenous dicarboxylic acid (such as glutarate, ketoglutarate, etc.) is simultaneously transported out of the cell.[5] As a result of the constant removal of endogenous dicarboxylic acid, OAT1-positive cells are at risk of depleting their supply of dicarboxylates. Once the supply of dicarboxylates is depleted, the OAT1 transporter can no longer function.
To prevent the loss of endogenous dicarboxylates, OAT1-positive cells also express a sodium-dicarboxylate cotransporter called NaDC3 that transports dicarboxylates back into the OAT1-positive cell. Sodium is required to drive this process. In the absence of a sodium gradient across the cell membrane, the NaDC3 cotransporter ceases to function, intra-cellular dicarboxylates are depleted, and the OAT1 transporter also grinds to a halt.[10]
The renal organic anion transporters OAT1, OAT3, OATP4C1, MDR1, MRP2, MRP4 and URAT1 are expressed in the S2 segment of the proximal convoluted tubules of the kidneys. OAT1, OAT3, and OATP4C1 transport small organic anions from the plasma into the S2 cells. MDR1, MRP2, MRP4 and URAT1 then transports these organic anions from the cytoplasm of the S2 cells into the lumen of the proximal convoluted tubules. These organic anions are then excreted in the urine.[5]
Substrates
Known substrates of OAT1 include para-aminohippurate (PAH), dicarboxylates, prostaglandins, cyclic nucleotides, urate, folate, diuretics, ACE inhibitors, antiviral agents, beta-lactam antibiotics, antineoplastics, mycotoxins, sulfate conjugates, glucuronide conjugates, cysteine conjugates, ochratoxin A, NSAIDs, mercapturic acids and uremic toxins.[5]
Regulation
Alterations in the expression and function of OAT1 play important roles in intra- and inter-individual variability of the therapeutic efficacy and the toxicity of many drugs. As a result, the activity of OAT1 must be under tight regulation so as to carry out their normal functions.[11] The regulation of OAT transport activity in response to various stimuli can occur at several levels such as transcription, translation, and posttranslational modification. Posttranslational regulation is of particular interest, because it usually happens within a very short period of time (minutes to hours) when the body has to deal with rapidly changing amounts of substances as a consequence of variable intake of drugs, fluids, or meals as well as metabolic activity.[11] Post-translational modification is a process where new functional group(s) are conjugated to the amino acid side chains in a target protein through reversible or irreversible biochemical reactions. The common modifications include glycosylation, phosphorylation, ubiquitination,[11] sulfation, methylation, acetylation, and hydroxylation.
Antiviral induced Fanconi syndrome
Nucleoside analogs are a class of antiviral drugs that work by inhibiting viral nucleic acid synthesis. The nucleoside analogs acyclovir (ACV), zidovudine (AZT), didanosine (ddI), zalcitabine (ddC), lamivudine (3TC), stavudine (d4T), trifluridine,[12] cidofovir, adefovir,[13] and tenofovir (TDF) [14] are substrates of the OAT1 transporter. This may result in the buildup of these drugs in the proximal tubule cells. At high concentrations, these drugs inhibit DNA replication. This, in turn, may impair the function of these cells and may be the cause of antiviral induced Fanconi syndrome. The use of stavudine,[15] didenosine, abacavir, adefovir,[16] cidofovir [17] and tenofovir has been associated with Fanconi syndrome. Clinical features of tenofovir-induced Fanconi syndrome include glycosuria in the setting of normal serum glucose levels, phosphate wasting with hypophosphatemia, proteinuria (usually mild), acidosis, and hypokalemia, with or without acute renal failure.[18]
Mitochondrial inhibition
Since nucleoside analogs can build up in OAT1-positive cells and can inhibit mitochondrial replication, these drugs may lead to the depletion of mitochondria inside renal proximal tubules. Renal biopsies have demonstrated the depletion of tubule cell mitochondria among individuals receiving antiviral therapy with tenofovir. The remaining mitochondria were enlarged and dysmorphic.[19] In vitro the antiviral drugs didanosine and zidovudine are more potent inhibitors of mitochondrial DNA synthesis than tenofovir (ddI > AZT > TDF).[20] In its non-phosphorylated form, the drug acyclovir does not significantly inhibit mitochondrial DNA synthesis, unless the cell happens to be infected with a herpes virus.
Stavudine, zidovudine and indinavir (IDV) cause a decrease in mitochondrial respiration and an increase in mitochondrial mass in fat cells. Stavudine also causes severe mitochondrial DNA depletion. Combining zidovudine with stavudine does not increase the mitochondrial toxicity compared to stavudine alone. Both of these drugs must be phosphorylated by host enzymes before they become active. Zidovudine inhibits the phosphorylation of stavudine. This might reduce the toxicity of the combination. Using indinavir in combination with the other two drugs did not increase the toxicity of the combination. Indinavir is a protease inhibitor and works by a different mechanism than the other antiviral drugs. (d4T+AZT+IDV = d4T+AZT = d4T+IDV > AZT+IDV = AZT = IDV). All three of these drugs inhibit the expression of respiratory chain subunits (cytochrome c oxidase [CytOx]2 and CytOx4) in white fat cells but not brown fat cells.[21] Since stavudine and zidovudine are OAT1 substrates, they may have similar effects on proximal renal tubule cells as they do on fat cells.
Lamivudine has reverse chirality compared to didanosine, stavudine, zidovudine, and natural nucleosides. Mitochondrial DNA polymerase may not recognize it as a substrate. Lamivudine is not toxic to mitochondria in vivo.[22] Individuals who had been taking didanosine combined with stavudine exhibited improved mitochondrial function when they switched to lamivudine combined with tenofovir.[22][23]
Mitochondrial toxicity of OAT1 substrates:
- in vitro:
- d4T+AZT = d4T > AZT
- ddI > AZT > TDF > ACV
- in vivo
- d4T > AZT
- ddI > AZT > TDF
- d4T + ddI > 3TC + TDF
See also
References
- Sekine T, Cha SH, Endou H (July 2000). "The multispecific organic anion transporter (OAT) family" (PDF). Pflügers Arch. 440 (3): 337–50. doi:10.1007/s004240000297. PMID 10954321. S2CID 32469988.
- Reid G, Wolff NA, Dautzenberg FM, Burckhardt G (Jan 1999). "Cloning of a human renal p-aminohippurate transporter, hROAT1". Kidney Blood Press Res. 21 (2–4): 233–7. doi:10.1159/000025863. PMID 9762842. S2CID 46811285.
- Lu R, Chan BS, Schuster VL (Mar 1999). "Cloning of the human kidney PAH transporter: narrow substrate specificity and regulation by protein kinase C". Am J Physiol. 276 (2 Pt 2): F295–303. doi:10.1152/ajprenal.1999.276.2.F295. PMID 9950961.
- "Entrez Gene: SLC22A6 solute carrier family 22 (organic anion transporter), member 6".
- Sekine T, Miyazaki H, Endou H (February 2006). "Molecular physiology of renal organic anion transporters". Am. J. Physiol. Renal Physiol. 290 (2): F251–61. doi:10.1152/ajprenal.00439.2004. PMID 16403838.
- GRCh38: Ensembl release 89: ENSG00000197901 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000024650 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Stellmer F, Keyser B, Burckhardt BC, et al. (July 2007). "3-Hydroxyglutaric acid is transported via the sodium-dependent dicarboxylate transporter NaDC3". J. Mol. Med. 85 (7): 763–70. doi:10.1007/s00109-007-0174-5. PMID 17356845. S2CID 2922696.
- Xu D, Wang H, You G (2016). "Posttranslational Regulation of Organic Anion Transporters by Ubiquitination: Known and Novel". Med Res Rev. 36 (5): 964–79. doi:10.1002/med.21397. PMC 5147025. PMID 27291023.
- Wada S, Tsuda M, Sekine T, Cha SH, Kimura M, Kanai Y, Endou H (September 2000). "Rat multispecific organic anion transporter 1 (rOAT1) transports zidovudine, acyclovir, and other antiviral nucleoside analogs". J. Pharmacol. Exp. Ther. 294 (3): 844–9. PMID 10945832.
- Ho ES, Lin DC, Mendel DB, Cihlar T (March 2000). "Cytotoxicity of antiviral nucleotides adefovir and cidofovir is induced by the expression of human renal organic anion transporter 1". J. Am. Soc. Nephrol. 11 (3): 383–93. doi:10.1681/ASN.V113383. PMID 10703662.
- Kohler JJ; Hosseini SH; Green E; Russ R; Santoianni R; Lewis w (April 2010). "OAT1 Knock-out Mice Define Its Role in Tenofovir Transport and Renal Proximal Tubular Mitochondrial Toxicity". FASEB J. 24 (1_MeetingAbstracts): Meeting abstracts, 1030.1. doi:10.1096/fasebj.24.1_supplement.1030.1.
- Nelson M, Azwa A, Sokwala A, Harania RS, Stebbing J (2008). "Fanconi syndrome and lactic acidosis associated with stavudine and lamivudine therapy". AIDS. 22 (11): 1374–6. doi:10.1097/QAD.0b013e328303be50. PMID 18580619. S2CID 5229576.
- Ahmad M (2006). "Abacavir-induced reversible Fanconi syndrome with nephrogenic diabetes insipidus in a patient with acquired immunodeficiency syndrome". J Postgrad Med. 52 (4): 296–7. PMID 17102551.
- Vittecoq D, Dumitrescu L, Beaufils H, Deray G (August 1997). "Fanconi syndrome associated with cidofovir therapy". Antimicrob. Agents Chemother. 41 (8): 1846. doi:10.1128/AAC.41.8.1846. PMC 164022. PMID 9257778.
- Atta MG, Fine DM (March 2009). "Editorial comment: tenofovir nephrotoxicity--the disconnect between clinical trials and real-world practice". AIDS Read. 19 (3): 118–9. PMID 19334329.
- Herlitz LC, Mohan S, Stokes MB, Radhakrishnan J, D'Agati VD, Markowitz GS (September 2010). "Tenofovir nephrotoxicity: acute tubular necrosis with distinctive clinical, pathological, and mitochondrial abnormalities". Kidney Int. 78 (11): 1171–1177. doi:10.1038/ki.2010.318. PMID 20811330.
- Vidal F, Domingo JC, Guallar J, et al. (November 2006). "In vitro cytotoxicity and mitochondrial toxicity of tenofovir alone and in combination with other antiretrovirals in human renal proximal tubule cells". Antimicrob. Agents Chemother. 50 (11): 3824–32. doi:10.1128/AAC.00437-06. PMC 1635212. PMID 16940060.
- Viengchareun S, Caron M, Auclair M, et al. (2007). "Mitochondrial toxicity of indinavir, stavudine and zidovudine involves multiple cellular targets in white and brown adipocytes". Antivir. Ther. (Lond.). 12 (6): 919–29. doi:10.1177/135965350701200610. PMID 17926646. S2CID 25419054.
- Honkoop P, de Man RA, Scholte HR, Zondervan PE, Van Den Berg JW, Rademakers LH, et al. (1997). "Effect of lamivudine on morphology and function of mitochondria in patients with chronic hepatitis B." Hepatology. 26 (1): 211–5. doi:10.1002/hep.510260128. PMID 9214472. S2CID 8029309.
- Ananworanich J, Nuesch R, Côté HC, Kerr SJ, Hill A, Jupimai T, et al. (2008). "Changes in metabolic toxicity after switching from stavudine/didanosine to tenofovir/lamivudine--a Staccato trial substudy". J Antimicrob Chemother. 61 (6): 1340–3. doi:10.1093/jac/dkn097. PMID 18339636.
Further reading
- Hosoyamada M, Sekine T, Kanai Y, Endou H (1999). "Molecular cloning and functional expression of a multispecific organic anion transporter from human kidney". Am. J. Physiol. 276 (1 Pt 2): F122–8. doi:10.1152/ajprenal.1999.276.1.F122. PMID 9887087.
- Race JE, Grassl SM, Williams WJ, Holtzman EJ (1999). "Molecular cloning and characterization of two novel human renal organic anion transporters (hOAT1 and hOAT3)". Biochem. Biophys. Res. Commun. 255 (2): 508–14. doi:10.1006/bbrc.1998.9978. PMID 10049739.
- Cihlar T, Lin DC, Pritchard JB, et al. (1999). "The antiviral nucleotide analogs cidofovir and adefovir are novel substrates for human and rat renal organic anion transporter 1". Mol. Pharmacol. 56 (3): 570–80. doi:10.1124/mol.56.3.570. PMID 10462545.
- Bahn A, Prawitt D, Buttler D, et al. (2000). "Genomic structure and in vivo expression of the human organic anion transporter 1 (hOAT1) gene". Biochem. Biophys. Res. Commun. 275 (2): 623–30. doi:10.1006/bbrc.2000.3230. PMID 10964714.
- Babu E, Takeda M, Narikawa S, et al. (2002). "Human organic anion transporters mediate the transport of tetracycline". Jpn. J. Pharmacol. 88 (1): 69–76. doi:10.1254/jjp.88.69. PMID 11855680.
- Burckhardt BC, Brai S, Wallis S, et al. (2003). "Transport of cimetidine by flounder and human renal organic anion transporter 1". Am. J. Physiol. Renal Physiol. 284 (3): F503–9. doi:10.1152/ajprenal.00290.2002. PMID 12429554.
- Ichida K, Hosoyamada M, Kimura H, et al. (2004). "Urate transport via human PAH transporter hOAT1 and its gene structure". Kidney Int. 63 (1): 143–55. doi:10.1046/j.1523-1755.2003.00710.x. PMID 12472777.
- Strausberg RL, Feingold EA, Grouse LH, et al. (2003). "Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences". Proc. Natl. Acad. Sci. U.S.A. 99 (26): 16899–903. Bibcode:2002PNAS...9916899M. doi:10.1073/pnas.242603899. PMC 139241. PMID 12477932.
- Wolff NA, Thies K, Kuhnke N, et al. (2004). "Protein kinase C activation downregulates human organic anion transporter 1-mediated transport through carrier internalization". J. Am. Soc. Nephrol. 14 (8): 1959–68. doi:10.1097/01.ASN.0000079040.55124.25. PMID 12874449.
- Sauvant C, Hesse D, Holzinger H, et al. (2004). "Action of EGF and PGE2 on basolateral organic anion uptake in rabbit proximal renal tubules and hOAT1 expressed in human kidney epithelial cells". Am. J. Physiol. Renal Physiol. 286 (4): F774–83. doi:10.1152/ajprenal.00326.2003. PMID 14644751. S2CID 7610758.
- Ota T, Suzuki Y, Nishikawa T, et al. (2004). "Complete sequencing and characterization of 21,243 full-length human cDNAs". Nat. Genet. 36 (1): 40–5. doi:10.1038/ng1285. PMID 14702039.
- Tanaka K, Xu W, Zhou F, You G (2004). "Role of glycosylation in the organic anion transporter OAT1". J. Biol. Chem. 279 (15): 14961–6. doi:10.1074/jbc.M400197200. PMID 14749323.
- Sakurai Y, Motohashi H, Ueo H, et al. (2004). "Expression levels of renal organic anion transporters (OATs) and their correlation with anionic drug excretion in patients with renal diseases". Pharm. Res. 21 (1): 61–7. doi:10.1023/B:PHAM.0000012153.71993.cb. hdl:2433/144752. PMID 14984259. S2CID 28592120.
- Bahn A, Ebbinghaus C, Ebbinghaus D, et al. (2005). "Expression studies and functional characterization of renal human organic anion transporter 1 isoforms". Drug Metab. Dispos. 32 (4): 424–30. doi:10.1124/dmd.32.4.424. PMID 15039295.
- Hong M, Zhou F, You G (2004). "Critical amino acid residues in transmembrane domain 1 of the human organic anion transporter hOAT1". J. Biol. Chem. 279 (30): 31478–82. doi:10.1074/jbc.M404686200. PMID 15145940.
- Zalups RK, Aslamkhan AG, Ahmad S (2005). "Human organic anion transporter 1 mediates cellular uptake of cysteine-S conjugates of inorganic mercury". Kidney Int. 66 (1): 251–61. doi:10.1111/j.1523-1755.2004.00726.x. PMID 15200431.
- Zalups RK, Ahmad S (2004). "Homocysteine and the renal epithelial transport and toxicity of inorganic mercury: role of basolateral transporter organic anion transporter 1". J. Am. Soc. Nephrol. 15 (8): 2023–31. doi:10.1097/01.ASN.0000135115.63412.A9. PMID 15284288.
- Fujita T, Brown C, Carlson EJ, et al. (2005). "Functional analysis of polymorphisms in the organic anion transporter, SLC22A6 (OAT1)". Pharmacogenet. Genomics. 15 (4): 201–9. doi:10.1097/01213011-200504000-00003. PMID 15864112. S2CID 24148270.