Otocephaly
Otocephaly, also known as agnathia–otocephaly complex, is a very rare and lethal cephalic disorder characterized by the absence of the mandible (agnathia), with the ears fused together just below the chin (synotia). It is caused by a disruption to the development of the first branchial arch. It occurs in every 1 in 70,000 embryos.
| Otocephaly | |
|---|---|
| Other names | Agnathia-otocephaly complex,[1] dysgnathia complex,[1] holoprosencephaly–agnathia,[1] |
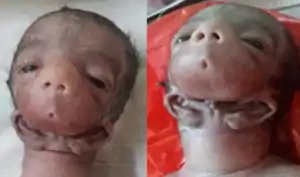 | |
| Female infant with otocephaly | |
| Specialty | Medical genetics |
| Symptoms | Absence of mandible (agnathia), small or absent mouth (microstomia), fused ears below chin (synotia), holoprosencephaly |
| Usual onset | 23rd–26th day of gestation (Carnegie stage 10) |
| Causes | Genetic |
| Diagnostic method | Prenatal ultrasound |
| Differential diagnosis | Treacher Collins syndrome, Goldenhar syndrome, Möbius syndrome |
| Prognosis | Stillbirth or miscarriage |
| Frequency | 1:70,000 |
Signs and symptoms



The disorder is characterised by the absence of the mandible (agnathia), with the ears fused together just below the chin (synotia). In addition to agnathia and synotia, other symptoms that may manifest in otocephaly include:[2]
- Facial/musculoskeletal
- Small (microglossia) or absent (aglossia) tongue
- Small (microstomia) or absent (astomia) mouth
- Cyclopia with proboscis
- Median cleft lip and cleft palate
- Neurological
- Organ systems
- Situs inversus (full rotation of the internal organs)
- Cardiac anomalies
- Ambiguous genitalia
- Absence of glands
Grades
Sewall Wright described twelve grades of otocephaly in guinea pigs.[3] Grades 1 to 5 were isolated agnathia with no neurological defects. Grades 6 to 9 featured severe holoprosencephaly. Grades 10 to 12 featured aprosopus (absence of the face and most of the head) with absence of the prosencephalon and mesencephalon. One human case corresponding to a high Wright grade has been reported in modern history.[4]
Cause
Otocephaly is generally a result of a de novo mutation in the gene PRRX1 on the long arm of chromosome 1.[1] Autosomal trisomies, while prevalent in related conditions like cyclopia, are uncommon in otocephaly.
Development
During early embryogenesis, many different organ systems begin development. Any disruption in these processes results in complex malformation, which usually results in death. The first branchial arch will normally develop around the 23rd to 26th day of gestation, also known as Carnegie stage 10. Usually, failure of this will result in isolated agnathia, but otocephaly may occur in exceptional circumstances. After agenesis of the first branchial arch, no cure is possible.
Prognosis
Otocephaly describes a spectrum of various manifestations, ranging in severity from severe micrognathia as a part of the Pierre Robin sequence to the cyclopia-holoprosencephaly complex, which is invariably associated with fetal death or death immediately after birth. Otocephaly is classified into four groups: (1) agnathia alone; (2) agnathia with holoprosencephaly; (3) agnathia with situs inversus and visceral anomalies; and (4) agnathia with holoprosencephaly, situs inversus, and visceral anomalies. Other extracranial malformations include neural tube defects, cephalocele, corpus callosum dysgenesis, renal ectopia, adrenal hypoplasia and rib, vertebral, or cardiac abnormalities.[5]
Because of the wide range of malformations, outcomes are variable. The most severe cases, often called cyclopia-holoprosencephaly complex, are almost invariably lethal, and patients show intrauterine growth restriction, prematurity, and impaired ventilation. Endotracheal intubation is difficult due to severe airway malformations, and only approximately 7 non-holoprosencephaly patients have survived beyond infancy.[6]
However, the less severe agnathia-otocephaly complex (AOC) has been successfully treated by extensive reconstructive surgery, including reconstruction of the mandible. Research indicates that a reasonable quality of life may even be achieved. However, the lack of musculature cannot be repaired with current therapies. This means swallowing is virtually impossible, so patients will depend on enteral nutrition. Researchers are optimistic that transplantation may eventually allow more complete reconstruction of structures affected by AOC.[7]
In all cases, an expert team, family information, premature birth planning, early gastrostomy and tracheostomy, and a long-term treatment plan are essential to ensure proper patient management.[8]
History
Otocephaly was first described in 1717 by Dutch scientist Theodor Kerckring. In 1933, evolutionary biologist Sewall Wright performed a study on otocephaly on guinea pigs and gave otocephaly its name.[3] The name comes from the Greek-derived Neo-Latin prefix oto- ("ear") and the suffix -cephaly ("head").
In 2018, Indian neonatologist Kanwar Singh and his associates described a particularly severe case of otocephaly with cyclopia, agnathia, complete astomia and synotia. They dubbed the resulting condition Kanwar syndrome.[2]
References
- "OMIM Entry - # 202650 - AGNATHIA-OTOCEPHALY COMPLEX; AGOTC". Online Mendelian Inheritance in Man. Retrieved 2019-06-05.
- Singh K, Sharma S, Agarwal K, Kalra A (2018). "Cyclopia-otocephaly-agnathia-synotia-astomia complex: A case report". Journal of Clinical Neonatology. 7 (3): 177. doi:10.4103/jcn.jcn_23_18. ISSN 2249-4847.
- Wright S (November 1934). "On the Genetics of Subnormal Development of the Head (Otocephaly) in the Guinea Pig". Genetics. 19 (6): 471–505. doi:10.1093/genetics/19.6.471. PMC 1208510. PMID 17246734.
- Utkus A, Kazakevicius R, Ptasekas R, Kucinskas V, Beckwith JB, Opitz JM (June 2001). "Human anotocephaly (aprosopus, acrania-synotia) in the Vilnius anatomical collection". American Journal of Medical Genetics. 101 (2): 163–171. doi:10.1002/ajmg.1320. PMID 11391661.
- Jagtap SV, Saini N, Jagtap S, Saini S (September 2015). "Otocephaly: Agnathia- Microstomia-Synotia Syndrome- A Rare Congenital Anomaly". Journal of Clinical and Diagnostic Research. 9 (9): ED03–ED04. doi:10.7860/JCDR/2015/13636.6444. PMC 4606241. PMID 26500912.
- Hisaba WJ, Milani HJ, Araujo Júnior E, Passos JP, Barreto EQ, Carvalho NS, et al. (December 2014). "Agnathia-otocephaly: prenatal diagnosis by two- and three-dimensional ultrasound and magnetic resonance imaging. Case report". Medical Ultrasonography. 16 (4): 377–379. doi:10.11152/mu.201.3.2066.164.wjh1. PMID 25463893.
- Golinko MS, Shetye P, Flores RL, Staffenberg DA (November 2015). "Severe Agnathia-Otocephaly Complex: Surgical Management and Longitudinal Follow-up From Birth Through Adulthood". The Journal of Craniofacial Surgery. 26 (8): 2387–2392. doi:10.1097/SCS.0000000000002150. PMID 26517463. S2CID 12250046.
- Díaz Del Arco C, Oliva A, Pelayo Alarcón A (Jan 2020). "Agnathia-microstomia-synotia syndrome (otocephaly)". Autopsy & Case Reports. 10 (1): e2020152. doi:10.4322/acr.2020.152. PMC 7059210. PMID 32185147.