Phosphoenolpyruvate mutase
In enzymology, a phosphoenolpyruvate mutase (EC 5.4.2.9) is an enzyme that catalyzes the chemical reaction
- phosphoenolpyruvate 3-phosphonopyruvate

| phosphoenolpyruvate mutase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| EC no. | 5.4.2.9 | ||||||||
| CAS no. | 115756-49-5 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
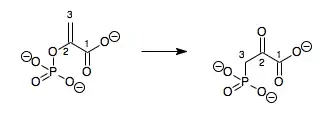
Hence, this enzyme has one substrate, phosphoenolpyruvate (PEP), and one product, 3-phosphonopyruvate (PPR), which are structural isomers.
This enzyme belongs to the family of isomerases, specifically the phosphotransferases (phosphomutases), which transfer phosphate groups within a molecule. The systematic name of this enzyme class is phosphoenolpyruvate 2,3-phosphonomutase. Other names in common use include phosphoenolpyruvate-phosphonopyruvate phosphomutase, PEP phosphomutase, phosphoenolpyruvate phosphomutase, PEPPM, and PEP phosphomutase. This enzyme participates in aminophosphonate metabolism.
Structural studies
As of late 2007, 6 structures have been solved for this class of enzymes, all by the Herzberg group at the University of Maryland using PEPPM from the blue mussel, Mytilus edulis. The first structure (PDB accession code 1PYM) was solved in 1999 and featured a magnesium oxalate inhibitor.[3] This structure identified the enzyme as consisting of identical beta barrel subunits (exhibiting the TIM barrel fold, which consists of eight parallel beta strands). Dimerization was observed in which a helix from each subunit interacts with the other subunit's barrel; the authors called this feature "helix swapping." The dimers can dimerize as well to form a homotetrameric enzyme. A double phosphoryl transfer mechanism was proposed on the basis of this study: this would involve breakage of PEP's phosphorus-oxygen bond to form a phosphoenzyme intermediate, followed by transfer of the phosphoryl group from the enzyme to carbon-3, forming PPR.
However, more recently, a structure with a sulfopyruvate inhibitor, which is a closer substrate analogue, was solved (1M1B);[4] this study supported instead a dissociative mechanism. A notable feature of these structures was the shielding of the active site from solvent; it was proposed that a significant conformational change takes place on binding to allow this, moving the protein from an "open" to a "closed" state, and this was supported by several crystal structures in the open state.[5] Three of these were of the wild type: the apoenzyme in 1S2T, the enzyme plus its magnesium ion cofactor in 1S2V, and the enzyme at high ionic strength in 1S2W. A mutant (D58A, in one of the active-site loops) was crystallized as an apoenzyme also (1S2U). From these structures, an active-site "gating" loop (residues 115-133) that shields the substrate from solvent in the closed conformation was identified.
The two conformations, taken from the crystal structures 1M1B (closed) and 1S2T (open), are docked into each other in the images below; they differ negligibly except in the gating loop, which is colored purple for the closed conformation and blue for the open conformation. In the active-site closeup (left), several sidechains (cyan) that have been identified as important in catalysis are included as well; the overview (right) illustrates the distinctive helix-swapping fold. The images are still shots from ribbon kinemages. Both of these structures were crystallized as dimers. In chain A (used for the active-site closeup), helices are red while loops (other than the gating loop) are white and beta strands are green; in chain B, helices are yellow, beta strands are olive, and loops are gray; these colors are the same for the closed and open structures. Magnesium ions are gray and the sulfopyruvate ligands are pink; both are from the closed structure (though the enzyme has also been crystallized with only magnesium bound, and it adopted an open conformation).
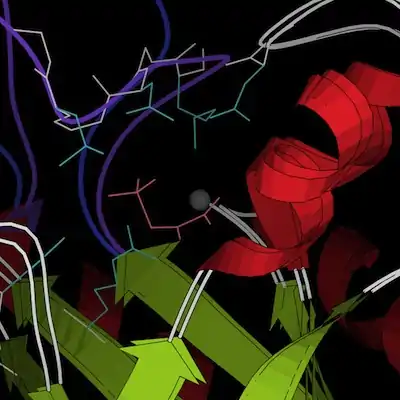
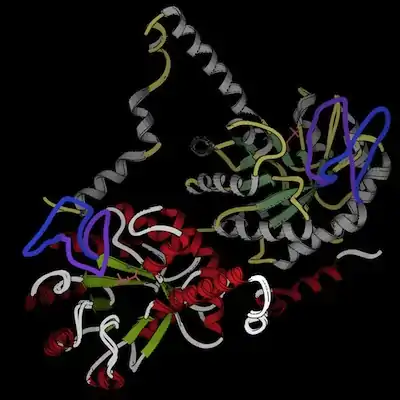
The structure of PEPPM is very similar to that of methylisocitrate lyase, an enzyme involved in propanoate metabolism whose substrate is also a low-molecular weight carboxylic acid—the beta-barrel structure as well as the active site layout and multimerization geometry are the same. Isocitrate lyase is also quite similar, though each subunit has a second, smaller beta domain in addition to the main beta barrel.
Mechanism
Phosphoenolpyruvate mutase is thought to exhibit a dissociative mechanism.[4] A magnesium ion is involved as a cofactor. The phosphoryl/phosphate group also appears to interact ionically with Arg159 and His190, stabilizing the reactive intermediate. A phosphoenzyme intermediate is unlikely because the most feasible residues for the covalent adduct can be mutated with only partial loss of function. The reaction involves dissociation of phosphorus from oxygen 2 and then a nucleophilic attack by carbon 3 on phosphorus. Notably, the configuration is retained at phosphorus, i.e. carbon 3 of PPR adds to the same face of phosphorus from which oxygen 2 of PEP was removed; this would be unlikely for a non-enzyme-catalyzed dissociative mechanism, but since the reactive intermediate interacts strongly with the amino acids and magnesium ions of the active site, it is to be expected in the presence of enzyme catalysis.
Residues in the active-site gating loop, particularly Lys120, Asn122, and Leu124, also appear to interact with the substrate and reactive intermediate; these interactions explain why the loop moves into the closed conformation on substrate binding.
Biological function
Because phosphoenolpyruvate mutase has the unusual ability to form a new carbon-phosphorus bond, it is essential to the synthesis of phosphonates, such as phosphonolipids and the antibiotics fosfomycin and bialaphos. The formation of this bond is quite thermodynamically unfavorable; even though PEP is a very high-energy phosphate compound, the equilibrium in PEP-PPR interconversion still favors PEP.[1] The enzyme phosphonopyruvate decarboxylase presents a solution to this problem: it catalyzes the very thermodynamically favorable decarboxylation of PPR, and the resulting 2-phosphonoacetaldehyde is then converted into biologically useful phosphonates. This allows phosphoneolpyruvate's reaction to proceed in the forward direction, due to Le Chatelier's principle. The decarboxylation removes product quickly, and thus the reaction moves forward even though there would be much more reactant than product if the system were allowed to reach equilibrium by itself.
The enzyme carboxyphosphoenolpyruvate phosphonomutase performs a similar reaction, converting P-carboxyphosphoenolpyruvate to phosphinopyruvate and carbon dioxide. [6]
References
- Bowman E, McQueney M, Barry RJ, Dunaway-Mariano D (1988). "Catalysis and thermodynamics of the phosphoenolpyruvate phosphonopyruvate rearrangement - entry into the phosphonate class of naturally-occurring organo-phosphorus compounds". J. Am. Chem. Soc. 110 (16): 5575–5576. doi:10.1021/ja00224a054.
- Seidel HM, Freeman S, Seto H, Knowles JR (1988). "Phosphonate biosynthesis: isolation of the enzyme responsible for the formation of a carbon-phosphorus bond". Nature. 335 (6189): 457–458. Bibcode:1988Natur.335..457S. doi:10.1038/335457a0. PMID 3138545. S2CID 4310660.
- Huang K, Li Z, Jia Y, Dunaway-Mariano D, Herzberg O (1999). "Helix swapping between two alpha/beta barrels: crystal structure of phosphoenolpyruvate mutase with bound Mg(2+)-oxalate". Struct. Fold. Des. 7 (5): 539–48. doi:10.1016/S0969-2126(99)80070-7. PMID 10378273.
- Liu S, Lu Z, Jia Y, Dunaway-Mariano D, Herzberg O (2002). "Dissociative phosphoryl transfer in PEP mutase catalysis: structure of the enzyme/sulfopyruvate complex and kinetic properties of mutants". Biochemistry. 41 (32): 10270–10276. doi:10.1021/bi026024v. PMID 12162742.
- Liu S, Lu Z, Han Y, Jia Y, Howard A, Dunaway-Mariano D, Herzberg O (2004). "Conformational flexibility of PEP mutase". Biochemistry. 43 (15): 4447–4453. CiteSeerX 10.1.1.432.6514. doi:10.1021/bi036255h. PMID 15078090.
- Hidaka T, Imai S, Hara O, Anzai H, Murakami T, Nagaoka K, Seto H (1990). "Carboxyphosphonoenolpyruvate phosphonomutase, a novel enzyme catalyzing C-P bond formation". J. Bacteriol. 172 (6): 3066–72. doi:10.1128/jb.172.6.3066-3072.1990. PMC 209109. PMID 2160937.