Proteinopathy
In medicine, proteinopathy ([pref. protein]; -pathy [suff. disease]; proteinopathies pl.; proteinopathic adj), or proteopathy, protein conformational disorder, or protein misfolding disease, is a class of diseases in which certain proteins become structurally abnormal, and thereby disrupt the function of cells, tissues and organs of the body.[1][2] Often the proteins fail to fold into their normal configuration; in this misfolded state, the proteins can become toxic in some way (a toxic gain-of-function) or they can lose their normal function.[3] The proteinopathies include such diseases as Creutzfeldt–Jakob disease and other prion diseases, Alzheimer's disease, Parkinson's disease, amyloidosis, multiple system atrophy, and a wide range of other disorders.[2][4][5][6][7][8] The term proteopathy was first proposed in 2000 by Lary Walker and Harry LeVine.[1]
| Proteinopathy | |
|---|---|
 | |
| Micrograph of a section of the cerebral cortex from a person with Alzheimer's disease, immunostained with an antibody to amyloid beta (brown), a protein fragment that accumulates in amyloid plaques and cerebral amyloid angiopathy. 10X microscope objective. |
The concept of proteopathy can trace its origins to the mid-19th century, when, in 1854, Rudolf Virchow coined the term amyloid ("starch-like") to describe a substance in cerebral corpora amylacea that exhibited a chemical reaction resembling that of cellulose. In 1859, Friedreich and Kekulé demonstrated that, rather than consisting of cellulose, "amyloid" actually is rich in protein.[9] Subsequent research has shown that many different proteins can form amyloid, and that all amyloids show birefringence in cross-polarized light after staining with the dye Congo red, as well as a fibrillar ultrastructure when viewed with an electron microscope.[9] However, some proteinaceous lesions lack birefringence and contain few or no classical amyloid fibrils, such as the diffuse deposits of amyloid beta (Aβ) protein in the brains of people with Alzheimer's.[10] Furthermore, evidence has emerged that small, non-fibrillar protein aggregates known as oligomers are toxic to the cells of an affected organ, and that amyloidogenic proteins in their fibrillar form may be relatively benign.[11][12]
Pathophysiology
In most, if not all proteinopathies, a change in the 3-dimensional folding conformation increases the tendency of a specific protein to bind to itself.[5] In this aggregated form, the protein is resistant to clearance and can interfere with the normal capacity of the affected organs. In some cases, misfolding of the protein results in a loss of its usual function. For example, cystic fibrosis is caused by a defective cystic fibrosis transmembrane conductance regulator (CFTR) protein,[3] and in amyotrophic lateral sclerosis / frontotemporal lobar degeneration (FTLD), certain gene-regulating proteins inappropriately aggregate in the cytoplasm, and thus are unable to perform their normal tasks within the nucleus.[13][14] Because proteins share a common structural feature known as the polypeptide backbone, all proteins have the potential to misfold under some circumstances.[15] However, only a relatively small number of proteins are linked to proteopathic disorders, possibly due to structural idiosyncrasies of the vulnerable proteins. For example, proteins that are normally unfolded or relatively unstable as monomers (that is, as single, unbound protein molecules) are more likely to misfold into an abnormal conformation.[5][15][16] In nearly all instances, the disease-causing molecular configuration involves an increase in beta-sheet secondary structure of the protein.[5][15][17][18][19] The abnormal proteins in some proteopathies have been shown to fold into multiple 3-dimensional shapes; these variant, proteinaceous structures are defined by their different pathogenic, biochemical, and conformational properties.[20] They have been most thoroughly studied with regard to prion disease, and are referred to as protein strains.[21][22]
_of_alpha-synuclein_in_Lewy_Bodies_and_Lewy_Neurites_in_the_neocortex_of_a_patient_with_Lewy_Body_Disease.jpg.webp)
The likelihood that proteinopathy will develop is increased by certain risk factors that promote the self-assembly of a protein. These include destabilizing changes in the primary amino acid sequence of the protein, post-translational modifications (such as hyperphosphorylation), changes in temperature or pH, an increase in production of a protein, or a decrease in its clearance.[1][5][15] Advancing age is a strong risk factor,[1] as is traumatic brain injury.[23][24] In the aging brain, multiple proteopathies can overlap.[25] For example, in addition to tauopathy and Aβ-amyloidosis (which coexist as key pathologic features of Alzheimer's disease), many Alzheimer patients have concomitant synucleinopathy (Lewy bodies) in the brain.[26]
It is hypothesized that chaperones and co-chaperones (proteins that assist protein folding) may antagonize proteotoxicity during aging and in protein misfolding-diseases to maintain proteostasis.[27][28][29]
Seeded induction
Some proteins can be induced to form abnormal assemblies by exposure to the same (or similar) protein that has folded into a disease-causing conformation, a process called 'seeding' or 'permissive templating'.[30][31] In this way, the disease state can be brought about in a susceptible host by the introduction of diseased tissue extract from an affected donor. The best known forms of inducible proteopathy are prion diseases,[32] which can be transmitted by exposure of a host organism to purified prion protein in a disease-causing conformation.[33][34] There is now evidence that other proteinopathies can be induced by a similar mechanism, including Aβ amyloidosis, amyloid A (AA) amyloidosis, and apolipoprotein AII amyloidosis,[31][35] tauopathy,[36] synucleinopathy,[37][38][39][40] and the aggregation of superoxide dismutase-1 (SOD1),[41][42] polyglutamine,[43][44] and TAR DNA-binding protein-43 (TDP-43).[45]
In all of these instances, an aberrant form of the protein itself appears to be the pathogenic agent. In some cases, the deposition of one type of protein can be experimentally induced by aggregated assemblies of other proteins that are rich in β-sheet structure, possibly because of structural complementarity of the protein molecules. For example, AA amyloidosis can be stimulated in mice by such diverse macromolecules as silk, the yeast amyloid Sup35, and curli fibrils from the bacterium Escherichia coli.[46] AII amyloid can be induced in mice by a variety of β-sheet rich amyloid fibrils,[47] and cerebral tauopathy can be induced by brain extracts that are rich in aggregated Aβ.[48] There is also experimental evidence for cross-seeding between prion protein and Aβ.[49] In general, such heterologous seeding is less efficient than is seeding by a corrupted form of the same protein.
List of proteinopathies
Management
The development of effective treatments for many proteopathies has been challenging.[75][76] Because the proteopathies often involve different proteins arising from different sources, treatment strategies must be customized to each disorder; however, general therapeutic approaches include maintaining the function of affected organs, reducing the formation of the disease-causing proteins, preventing the proteins from misfolding and/or aggregating, or promoting their removal.[77][75][78] For example, in Alzheimer's disease, researchers are seeking ways to reduce the production of the disease-associated protein Aβ by inhibiting the enzymes that free it from its parent protein.[76] Another strategy is to use antibodies to neutralize specific proteins by active or passive immunization.[79] In some proteopathies, inhibiting the toxic effects of protein oligomers might be beneficial.[80] Amyloid A (AA) amyloidosis can be reduced by treating the inflammatory state that increases the amount of the protein in the blood (referred to as serum amyloid A, or SAA).[75] In immunoglobulin light chain amyloidosis (AL amyloidosis), chemotherapy can be used to lower the number of the blood cells that make the light chain protein that forms amyloid in various bodily organs.[81] Transthyretin (TTR) amyloidosis (ATTR) results from the deposition of misfolded TTR in multiple organs.[82] Because TTR is mainly produced in the liver, TTR amyloidosis can be slowed in some hereditary cases by liver transplantation.[83] TTR amyloidosis also can be treated by stabilizing the normal assemblies of the protein (called tetramers because they consist of four TTR molecules bound together). Stabilization prevents individual TTR molecules from escaping, misfolding, and aggregating into amyloid.[84][85]
Several other treatment strategies for proteopathies are being investigated, including small molecules and biologic medicines such as small interfering RNAs, antisense oligonucleotides, peptides, and engineered immune cells.[84][81][86][87] In some cases, multiple therapeutic agents may be combined to improve effectiveness.[81][88]
Additional images
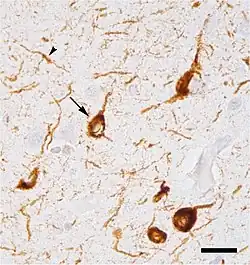 Micrograph of tauopathy (brown) in a neuronal cell body (arrow) and process (arrowhead) in the cerebral cortex of a patient with Alzheimer's disease. Bar = 25 microns (0.025mm).
Micrograph of tauopathy (brown) in a neuronal cell body (arrow) and process (arrowhead) in the cerebral cortex of a patient with Alzheimer's disease. Bar = 25 microns (0.025mm).
References
- Walker LC, LeVine H (2000). "The cerebral proteopathies". Neurobiology of Aging. 21 (4): 559–61. doi:10.1016/S0197-4580(00)00160-3. PMID 10924770. S2CID 54314137.
- Walker LC, LeVine H (2000). "The cerebral proteopathies: neurodegenerative disorders of protein conformation and assembly". Molecular Neurobiology. 21 (1–2): 83–95. doi:10.1385/MN:21:1-2:083. PMID 11327151. S2CID 32618330.
- Luheshi LM, Crowther DC, Dobson CM (February 2008). "Protein misfolding and disease: from the test tube to the organism". Current Opinion in Chemical Biology. 12 (1): 25–31. doi:10.1016/j.cbpa.2008.02.011. PMID 18295611.
- Chiti F, Dobson CM (2006). "Protein misfolding, functional amyloid, and human disease". Annual Review of Biochemistry. 75 (1): 333–66. doi:10.1146/annurev.biochem.75.101304.123901. PMID 16756495. S2CID 23797549.
- Carrell RW, Lomas DA (July 1997). "Conformational disease". Lancet. 350 (9071): 134–8. doi:10.1016/S0140-6736(97)02073-4. PMID 9228977. S2CID 39124185.
- Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, Masters CL, Merlini G, Saraiva MJ, Sipe JD (September 2007). "A primer of amyloid nomenclature". Amyloid. 14 (3): 179–83. doi:10.1080/13506120701460923. PMID 17701465. S2CID 12480248.
- Westermark GT, Fändrich M, Lundmark K, Westermark P (January 2018). "Noncerebral Amyloidoses: Aspects on Seeding, Cross-Seeding, and Transmission". Cold Spring Harbor Perspectives in Medicine. 8 (1): a024323. doi:10.1101/cshperspect.a024323. PMC 5749146. PMID 28108533.
- Prusiner SB (2013). "Biology and genetics of prions causing neurodegeneration". Annual Review of Genetics. 47: 601–23. doi:10.1146/annurev-genet-110711-155524. PMC 4010318. PMID 24274755.
- Sipe JD, Cohen AS (June 2000). "Review: history of the amyloid fibril". Journal of Structural Biology. 130 (2–3): 88–98. doi:10.1006/jsbi.2000.4221. PMID 10940217.
- Wisniewski HM, Sadowski M, Jakubowska-Sadowska K, Tarnawski M, Wegiel J (July 1998). "Diffuse, lake-like amyloid-beta deposits in the parvopyramidal layer of the presubiculum in Alzheimer disease". Journal of Neuropathology and Experimental Neurology. 57 (7): 674–83. doi:10.1097/00005072-199807000-00004. PMID 9690671.
- Glabe CG (April 2006). "Common mechanisms of amyloid oligomer pathogenesis in degenerative disease". Neurobiology of Aging. 27 (4): 570–5. doi:10.1016/j.neurobiolaging.2005.04.017. PMID 16481071. S2CID 32899741.
- Gadad BS, Britton GB, Rao KS (2011). "Targeting oligomers in neurodegenerative disorders: lessons from α-synuclein, tau, and amyloid-β peptide". Journal of Alzheimer's Disease. 24 (Suppl 2): 223–32. doi:10.3233/JAD-2011-110182. PMID 21460436.
- Ito D, Suzuki N (October 2011). "Conjoint pathologic cascades mediated by ALS/FTLD-U linked RNA-binding proteins TDP-43 and FUS". Neurology. 77 (17): 1636–43. doi:10.1212/WNL.0b013e3182343365. PMC 3198978. PMID 21956718.
- Wolozin B, Apicco D (2015). "RNA Binding Proteins and the Genesis of Neurodegenerative Diseases". GeNeDis 2014. Advances in Experimental Medicine and Biology. Vol. 822. pp. 11–5. doi:10.1007/978-3-319-08927-0_3. ISBN 978-3-319-08926-3. PMC 4694570. PMID 25416971.
- Dobson CM (September 1999). "Protein misfolding, evolution and disease". Trends in Biochemical Sciences. 24 (9): 329–32. doi:10.1016/S0968-0004(99)01445-0. PMID 10470028.
- Jucker M, Walker LC (September 2013). "Self-propagation of pathogenic protein aggregates in neurodegenerative diseases". Nature. 501 (7465): 45–51. Bibcode:2013Natur.501...45J. doi:10.1038/nature12481. PMC 3963807. PMID 24005412.
- Selkoe DJ (December 2003). "Folding proteins in fatal ways". Nature. 426 (6968): 900–4. Bibcode:2003Natur.426..900S. doi:10.1038/nature02264. PMID 14685251. S2CID 6451881.
- Eisenberg D, Jucker M (March 2012). "The amyloid state of proteins in human diseases". Cell. 148 (6): 1188–203. doi:10.1016/j.cell.2012.02.022. PMC 3353745. PMID 22424229.
- Röhr D, Boon BD (December 2020). "Label-free vibrational imaging of different Aβ plaque types in Alzheimer's disease reveals sequential events in plaque development". Acta Neuropathologica Communications. 8 (1): 222. doi:10.1186/s40478-020-01091-5. PMC 7733282. PMID 33308303.
- Walker LC (November 2016). "Proteopathic Strains and the Heterogeneity of Neurodegenerative Diseases". Annual Review of Genetics. 50: 329–346. doi:10.1146/annurev-genet-120215-034943. PMC 6690197. PMID 27893962.
- Collinge J, Clarke AR (November 2007). "A general model of prion strains and their pathogenicity". Science. 318 (5852): 930–6. Bibcode:2007Sci...318..930C. doi:10.1126/science.1138718. PMID 17991853. S2CID 8993435.
- Colby DW, Prusiner SB (September 2011). "De novo generation of prion strains". Nature Reviews. Microbiology. 9 (11): 771–7. doi:10.1038/nrmicro2650. PMC 3924856. PMID 21947062.
- DeKosky ST, Ikonomovic MD, Gandy S (September 2010). "Traumatic brain injury--football, warfare, and long-term effects". The New England Journal of Medicine. 363 (14): 1293–6. doi:10.1056/NEJMp1007051. PMID 20879875.
- McKee AC, Stein TD, Kiernan PT, Alvarez VE (May 2015). "The neuropathology of chronic traumatic encephalopathy". Brain Pathology. 25 (3): 350–64. doi:10.1111/bpa.12248. PMC 4526170. PMID 25904048.
- Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, Duyckaerts C, Frosch MP, Haroutunian V, Hof PR, Hulette CM, Hyman BT, Iwatsubo T, Jellinger KA, Jicha GA, Kövari E, Kukull WA, Leverenz JB, Love S, Mackenzie IR, Mann DM, Masliah E, McKee AC, Montine TJ, Morris JC, Schneider JA, Sonnen JA, Thal DR, Trojanowski JQ, Troncoso JC, Wisniewski T, Woltjer RL, Beach TG (May 2012). "Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature". Journal of Neuropathology and Experimental Neurology. 71 (5): 362–81. doi:10.1097/NEN.0b013e31825018f7. PMC 3560290. PMID 22487856.
- Mrak RE, Griffin WS (2007). "Dementia with Lewy bodies: Definition, diagnosis, and pathogenic relationship to Alzheimer's disease". Neuropsychiatric Disease and Treatment. 3 (5): 619–25. PMC 2656298. PMID 19300591.
- Douglas PM, Summers DW, Cyr DM (2009). "Molecular chaperones antagonize proteotoxicity by differentially modulating protein aggregation pathways". Prion. 3 (2): 51–8. doi:10.4161/pri.3.2.8587. PMC 2712599. PMID 19421006.
- Brehme M, Voisine C, Rolland T, Wachi S, Soper JH, Zhu Y, Orton K, Villella A, Garza D, Vidal M, Ge H, Morimoto RI (November 2014). "A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease". Cell Reports. 9 (3): 1135–50. doi:10.1016/j.celrep.2014.09.042. PMC 4255334. PMID 25437566.
- Brehme M, Voisine C (August 2016). "Model systems of protein-misfolding diseases reveal chaperone modifiers of proteotoxicity". Disease Models & Mechanisms. 9 (8): 823–38. doi:10.1242/dmm.024703. PMC 5007983. PMID 27491084.
- Hardy J (August 2005). "Expression of normal sequence pathogenic proteins for neurodegenerative disease contributes to disease risk: 'permissive templating' as a general mechanism underlying neurodegeneration". Biochemical Society Transactions. 33 (Pt 4): 578–81. doi:10.1042/BST0330578. PMID 16042548.
- Walker LC, Levine H, Mattson MP, Jucker M (August 2006). "Inducible proteopathies". Trends in Neurosciences. 29 (8): 438–43. doi:10.1016/j.tins.2006.06.010. PMID 16806508. S2CID 46630402.
- Prusiner SB (May 2001). "Shattuck lecture--neurodegenerative diseases and prions". The New England Journal of Medicine. 344 (20): 1516–26. doi:10.1056/NEJM200105173442006. PMID 11357156.
- Zou WQ, Gambetti P (April 2005). "From microbes to prions the final proof of the prion hypothesis". Cell. 121 (2): 155–7. doi:10.1016/j.cell.2005.04.002. PMID 15851020.
- Ma J (2012). "The role of cofactors in prion propagation and infectivity". PLOS Pathogens. 8 (4): e1002589. doi:10.1371/journal.ppat.1002589. PMC 3325206. PMID 22511864.
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M (September 2006). "Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host". Science. 313 (5794): 1781–4. Bibcode:2006Sci...313.1781M. doi:10.1126/science.1131864. PMID 16990547. S2CID 27127208.
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M, Tolnay M (July 2009). "Transmission and spreading of tauopathy in transgenic mouse brain". Nature Cell Biology. 11 (7): 909–13. doi:10.1038/ncb1901. PMC 2726961. PMID 19503072.
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ (August 2009). "Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein". Proceedings of the National Academy of Sciences of the United States of America. 106 (31): 13010–5. doi:10.1073/pnas.0903691106. PMC 2722313. PMID 19651612.
- Hansen C, Angot E, Bergström AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P (February 2011). "α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells". The Journal of Clinical Investigation. 121 (2): 715–25. doi:10.1172/JCI43366. PMC 3026723. PMID 21245577.
- Kordower JH, Dodiya HB, Kordower AM, Terpstra B, Paumier K, Madhavan L, Sortwell C, Steece-Collier K, Collier TJ (September 2011). "Transfer of host-derived α synuclein to grafted dopaminergic neurons in rat". Neurobiology of Disease. 43 (3): 552–7. doi:10.1016/j.nbd.2011.05.001. PMC 3430516. PMID 21600984.
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW (May 2008). "Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease". Nature Medicine. 14 (5): 504–6. doi:10.1038/nm1747. PMID 18391962. S2CID 11991816.
- Chia R, Tattum MH, Jones S, Collinge J, Fisher EM, Jackson GS (May 2010). Feany MB (ed.). "Superoxide dismutase 1 and tgSOD1 mouse spinal cord seed fibrils, suggesting a propagative cell death mechanism in amyotrophic lateral sclerosis". PLOS ONE. 5 (5): e10627. doi:10.1371/journal.pone.0010627. PMC 2869360. PMID 20498711.
- Münch C, O'Brien J, Bertolotti A (March 2011). "Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells". Proceedings of the National Academy of Sciences of the United States of America. 108 (9): 3548–53. Bibcode:2011PNAS..108.3548M. doi:10.1073/pnas.1017275108. PMC 3048161. PMID 21321227.
- Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR (February 2009). "Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates". Nature Cell Biology. 11 (2): 219–25. doi:10.1038/ncb1830. PMC 2757079. PMID 19151706.
- Pearce MM, Kopito RR (February 2018). "Prion-Like Characteristics of Polyglutamine-Containing Proteins". Cold Spring Harbor Perspectives in Medicine. 8 (2): a024257. doi:10.1101/cshperspect.a024257. PMC 5793740. PMID 28096245.
- Furukawa Y, Kaneko K, Watanabe S, Yamanaka K, Nukina N (May 2011). "A seeding reaction recapitulates intracellular formation of Sarkosyl-insoluble transactivation response element (TAR) DNA-binding protein-43 inclusions". The Journal of Biological Chemistry. 286 (21): 18664–72. doi:10.1074/jbc.M111.231209. PMC 3099683. PMID 21454603.
- Lundmark K, Westermark GT, Olsén A, Westermark P (April 2005). "Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism". Proceedings of the National Academy of Sciences of the United States of America. 102 (17): 6098–102. Bibcode:2005PNAS..102.6098L. doi:10.1073/pnas.0501814102. PMC 1087940. PMID 15829582.
- Fu X, Korenaga T, Fu L, Xing Y, Guo Z, Matsushita T, Hosokawa M, Naiki H, Baba S, Kawata Y, Ikeda S, Ishihara T, Mori M, Higuchi K (April 2004). "Induction of AApoAII amyloidosis by various heterogeneous amyloid fibrils". FEBS Letters. 563 (1–3): 179–84. doi:10.1016/S0014-5793(04)00295-9. PMID 15063745.
- Bolmont T, Clavaguera F, Meyer-Luehmann M, Herzig MC, Radde R, Staufenbiel M, Lewis J, Hutton M, Tolnay M, Jucker M (December 2007). "Induction of tau pathology by intracerebral infusion of amyloid-beta -containing brain extract and by amyloid-beta deposition in APP x Tau transgenic mice". The American Journal of Pathology. 171 (6): 2012–20. doi:10.2353/ajpath.2007.070403. PMC 2111123. PMID 18055549.
- Morales R, Estrada LD, Diaz-Espinoza R, Morales-Scheihing D, Jara MC, Castilla J, Soto C (March 2010). "Molecular cross talk between misfolded proteins in animal models of Alzheimer's and prion diseases". The Journal of Neuroscience. 30 (13): 4528–35. doi:10.1523/JNEUROSCI.5924-09.2010. PMC 2859074. PMID 20357103.
- Revesz T, Ghiso J, Lashley T, Plant G, Rostagno A, Frangione B, Holton JL (September 2003). "Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view". Journal of Neuropathology and Experimental Neurology. 62 (9): 885–98. doi:10.1093/jnen/62.9.885. PMID 14533778.
- Guo L, Salt TE, Luong V, Wood N, Cheung W, Maass A, Ferrari G, Russo-Marie F, Sillito AM, Cheetham ME, Moss SE, Fitzke FW, Cordeiro MF (August 2007). "Targeting amyloid-beta in glaucoma treatment". Proceedings of the National Academy of Sciences of the United States of America. 104 (33): 13444–9. Bibcode:2007PNAS..10413444G. doi:10.1073/pnas.0703707104. PMC 1940230. PMID 17684098.
- Prusiner, SB (2004). Prion Biology and Diseases (2 ed.). Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. ISBN 0-87969-693-1.
- Goedert M, Spillantini MG, Del Tredici K, Braak H (January 2013). "100 years of Lewy pathology". Nature Reviews. Neurology. 9 (1): 13–24. doi:10.1038/nrneurol.2012.242. PMID 23183883. S2CID 12590215.
- Clavaguera F, Hench J, Goedert M, Tolnay M (February 2015). "Invited review: Prion-like transmission and spreading of tau pathology". Neuropathology and Applied Neurobiology. 41 (1): 47–58. doi:10.1111/nan.12197. PMID 25399729. S2CID 45101893.
- Mann DM, Snowden JS (November 2017). "Frontotemporal lobar degeneration: Pathogenesis, pathology and pathways to phenotype". Brain Pathology. 27 (6): 723–736. doi:10.1111/bpa.12486. PMC 8029341. PMID 28100023.
- Grad LI, Fernando SM, Cashman NR (May 2015). "From molecule to molecule and cell to cell: prion-like mechanisms in amyotrophic lateral sclerosis". Neurobiology of Disease. 77: 257–65. doi:10.1016/j.nbd.2015.02.009. PMID 25701498. S2CID 18510138.
- Ludolph AC, Brettschneider J, Weishaupt JH (October 2012). "Amyotrophic lateral sclerosis". Current Opinion in Neurology. 25 (5): 530–5. doi:10.1097/WCO.0b013e328356d328. PMID 22918486.
- Orr HT, Zoghbi HY (July 2007). "Trinucleotide repeat disorders". Annual Review of Neuroscience. 30 (1): 575–621. doi:10.1146/annurev.neuro.29.051605.113042. PMID 17417937.
- Almeida B, Fernandes S, Abreu IA, Macedo-Ribeiro S (2013). "Trinucleotide repeats: a structural perspective". Frontiers in Neurology. 4: 76. doi:10.3389/fneur.2013.00076. PMC 3687200. PMID 23801983.
- Spinner NB (March 2000). "CADASIL: Notch signaling defect or protein accumulation problem?". The Journal of Clinical Investigation. 105 (5): 561–2. doi:10.1172/JCI9511. PMC 292459. PMID 10712425.
- Quinlan RA, Brenner M, Goldman JE, Messing A (June 2007). "GFAP and its role in Alexander disease". Experimental Cell Research. 313 (10): 2077–87. doi:10.1016/j.yexcr.2007.04.004. PMC 2702672. PMID 17498694.
- Ito D, Suzuki N (January 2009). "Seipinopathy: a novel endoplasmic reticulum stress-associated disease". Brain. 132 (Pt 1): 8–15. doi:10.1093/brain/awn216. PMID 18790819.
- Sipe JD, Benson MD, Buxbaum JN, Ikeda SI, Merlini G, Saraiva MJ, Westermark P (December 2016). "Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines". Amyloid. 23 (4): 209–213. doi:10.1080/13506129.2016.1257986. PMID 27884064.
- Lomas DA, Carrell RW (October 2002). "Serpinopathies and the conformational dementias". Nature Reviews Genetics. 3 (10): 759–68. doi:10.1038/nrg907. PMID 12360234. S2CID 21633779.
- Mukherjee A, Soto C (May 2017). "Prion-Like Protein Aggregates and Type 2 Diabetes". Cold Spring Harbor Perspectives in Medicine. 7 (5): a024315. doi:10.1101/cshperspect.a024315. PMC 5411686. PMID 28159831.
- Askanas V, Engel WK (January 2006). "Inclusion-body myositis: a myodegenerative conformational disorder associated with Abeta, protein misfolding, and proteasome inhibition". Neurology. 66 (2 Suppl 1): S39-48. doi:10.1212/01.wnl.0000192128.13875.1e. PMID 16432144. S2CID 24365234.
- Ecroyd H, Carver JA (January 2009). "Crystallin proteins and amyloid fibrils". Cellular and Molecular Life Sciences. 66 (1): 62–81. doi:10.1007/s00018-008-8327-4. PMID 18810322. S2CID 6580402. Archived from the original on 2018-07-23. Retrieved 2021-09-15.
- Surguchev A, Surguchov A (January 2010). "Conformational diseases: looking into the eyes". Brain Research Bulletin. 81 (1): 12–24. doi:10.1016/j.brainresbull.2009.09.015. PMID 19808079. S2CID 38832894.
- Huilgol SC, Ramnarain N, Carrington P, Leigh IM, Black MM (May 1998). "Cytokeratins in primary cutaneous amyloidosis". The Australasian Journal of Dermatology. 39 (2): 81–5. doi:10.1111/j.1440-0960.1998.tb01253.x. PMID 9611375. S2CID 25820489.
- Janig E, Stumptner C, Fuchsbichler A, Denk H, Zatloukal K (March 2005). "Interaction of stress proteins with misfolded keratins". European Journal of Cell Biology. 84 (2–3): 329–39. doi:10.1016/j.ejcb.2004.12.018. PMID 15819411.
- D'Souza A, Theis JD, Vrana JA, Dogan A (June 2014). "Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration". Amyloid. 21 (2): 71–5. doi:10.3109/13506129.2013.876984. PMC 4021035. PMID 24446896.
- Meng X, Clews J, Kargas V, Wang X, Ford RC (January 2017). "The cystic fibrosis transmembrane conductance regulator (CFTR) and its stability". Cellular and Molecular Life Sciences. 74 (1): 23–38. doi:10.1007/s00018-016-2386-8. PMC 5209436. PMID 27734094.
- Stuart MJ, Nagel RL (2004). "Sickle-cell disease". Lancet. 364 (9442): 1343–60. doi:10.1016/S0140-6736(04)17192-4. PMID 15474138. S2CID 8139305.
- Bernstein AM, Ritch R, Wolosin JM (July 2018). "Exfoliation syndrome: A disease of autophagy and LOXL1 proteopathy". Journal of Glaucoma. 27 (Supplement 1): S44–S53. doi:10.1097/IJG.0000000000000919. PMC 6028293. PMID 29547474.
- Pepys MB (2006). "Amyloidosis". Annu Rev Med. 57: 223–241. doi:10.1146/annurev.med.57.121304.131243. PMID 16409147.
- Holtzman DM, Morris JC, Goate AM (2011). "Alzheimer's disease: the challenge of the second century". Sci Transl Med. 3 (77): 77sr1. doi:10.1126/scitranslmed.3002369. PMC 3130546. PMID 21471435.
- Pepys MB (2001). "Pathogenesis, diagnosis and treatment of systemic amyloidosis". Phil Trans R Soc Lond B. 356 (1406): 203–211. doi:10.1098/rstb.2000.0766. PMC 1088426. PMID 11260801.
- Walker LC, LeVine H 3rd (2002). "Proteopathy: the next therapeutic frontier?". Curr Opin Investig Drugs. 3 (5): 782–7. PMID 12090553.
- Braczynski AK, Schulz JB, Bach JP (2017). "Vaccination strategies in tauopathies and synucleinopathies". J Neurochem. 143 (5): 467–488. doi:10.1111/jnc.14207. PMID 28869766.
- Klein WL (2013). "Synaptotoxic amyloid-β oligomers: a molecular basis for the cause, diagnosis, and treatment of Alzheimer's disease?". J Alzheimers Dis. 33 (Suppl 1): S49-65. doi:10.3233/JAD-2012-129039. PMID 22785404.
- Badar T, D'Souza A, Hari P (2018). "Recent advances in understanding and treating immunoglobulin light chain amyloidosis". F1000Res. 7: 1348. doi:10.12688/f1000research.15353.1. PMC 6117860. PMID 30228867.
- Carvalho A, Rocha A, Lobato L (2015). "Liver transplantation in transthyretin amyloidosis: issues and challenges". Liver Transpl. 21 (3): 282–292. doi:10.1002/lt.24058. PMID 25482846.
- Suhr OB, Herlenius G, Friman S, Ericzon BG (2000). "Liver transplantation for hereditary transthyretin amyloidosis". Liver Transpl. 6 (3): 263–276. doi:10.1053/lv.2000.6145. PMID 10827225.
- Suhr OB, Larsson M, Ericzon BG, Wilczek HE, et al. (2016). "Survival After Transplantation in Patients With Mutations Other Than Val30Met: Extracts From the FAP World Transplant Registry". Transplantation. 100 (2): 373–381. doi:10.1097/TP.0000000000001021. PMC 4732012. PMID 26656838.
- Coelho T, et al. (2016). "Mechanism of Action and Clinical Application of Tafamidis in Hereditary Transthyretin Amyloidosis". Neurol Ther. 5 (1): 1–25. doi:10.1007/s40120-016-0040-x. PMC 4919130. PMID 26894299.
- Yu D, et al. (2012). "Single-stranded RNAs use RNAi to potently and allele-selectively inhibit mutant huntingtin expression". Cell. 150 (5): 895–908. doi:10.1016/j.cell.2012.08.002. PMC 3444165. PMID 22939619.
- Nuvolone M, Merlini G (2017). "Emerging therapeutic targets currently under investigation for the treatment of systemic amyloidosis". Expert Opin Ther Targets. 21 (12): 1095–1110. doi:10.1080/14728222.2017.1398235. PMID 29076382. S2CID 46766370.
- Joseph NS, Kaufman JL (2018). "Novel Approaches for the Management of AL Amyloidosis". Curr Hematol Malig Rep. 13 (3): 212–219. doi:10.1007/s11899-018-0450-1. PMID 29951831. S2CID 49475930.