Hereditary spastic paraplegia
Hereditary spastic paraplegia (HSP) is a group of inherited diseases whose main feature is a progressive gait disorder. The disease presents with progressive stiffness (spasticity) and contraction in the lower limbs.[1] HSP is also known as hereditary spastic paraparesis, familial spastic paraplegia, French settlement disease, Strumpell disease, or Strumpell-Lorrain disease. The symptoms are a result of dysfunction of long axons in the spinal cord. The affected cells are the primary motor neurons; therefore, the disease is an upper motor neuron disease.[2] HSP is not a form of cerebral palsy even though it physically may appear and behave much the same as spastic diplegia. The origin of HSP is different from cerebral palsy. Despite this, some of the same anti-spasticity medications used in spastic cerebral palsy are sometimes used to treat HSP symptoms.
| Hereditary spastic paraplegia | |
|---|---|
| Specialty | Neurology |
HSP is caused by defects in transport of proteins, structural proteins, cell-maintaining proteins, lipids, and other substances through the cell. Long nerve fibers (axons) are affected because long distances make nerve cells particularly sensitive to defects in these mentioned mechanisms.[3][4]
The disease was first described in 1880 by the German neurologist Adolph Strümpell.[5] It was described more extensively in 1888 by Maurice Lorrain, a French physician.[6] Due to their contribution in describing the disease, it is still called Strümpell-Lorrain disease in French-speaking countries. The term hereditary spastic paraplegia was coined by Anita Harding in 1983.[7]
Signs and symptoms
Symptoms depend on the type of HSP inherited. The main feature of the disease is progressive spasticity in the lower limbs due to pyramidal tract dysfunction. This also results in brisk reflexes, extensor plantar reflexes, muscle weakness, and variable bladder disturbances. Furthermore, among the core symptoms of HSP are also included abnormal gait and difficulty in walking, decreased vibratory sense at the ankles, and paresthesia.[8] Individuals with HSP can experience extreme fatigue associated with central nervous system and neuromuscular disorders, which can be disabling.[9][10][11] Initial symptoms are typically difficulty with balance, stubbing the toe or stumbling. Symptoms of HSP may begin at any age, from infancy to older than 60 years. If symptoms begin during the teenage years or later, then spastic gait disturbance usually progresses over many years. Canes, walkers, and wheelchairs may eventually be required, although some people never require assistance devices.[12] Disability has been described as progressing more rapidly in adult onset forms.[13]
More specifically, patients with the autosomal dominant pure form of HSP reveal normal facial and extraocular movement. Although jaw jerk may be brisk in older subjects, there is no speech disturbance or difficulty of swallowing. Upper extremity muscle tone and strength are normal. In the lower extremities, muscle tone is increased at the hamstrings, quadriceps and ankles. Weakness is most notable at the iliopsoas, tibialis anterior, and to a lesser extent, hamstring muscles.[13] In the complex form of the disorder, additional symptoms are present. These include: peripheral neuropathy, amyotrophy, ataxia, intellectual disability, ichthyosis, epilepsy, optic neuropathy, dementia, deafness, or problems with speech, swallowing or breathing.[14]
Anita Harding[7] classified the HSP in a pure and complicated form. Pure HSP presents with spasticity in the lower limbs, associated with neurogenic bladder disturbance as well as lack of vibration sensitivity (pallhypesthesia). On the other hand, HSP is classified as complex when lower limb spasticity is combined with any additional neurological symptom.
This classification is subjective and patients with complex HSPs are sometimes diagnosed as having cerebellar ataxia with spasticity, intellectual disability (with spasticity), or leukodystrophy.[7] Some of the genes listed below have been described in other diseases than HSP before. Therefore, some key genes overlap with other disease groups.
Age of onset
In the past, HSP has been classified as early onset beginning in early childhood or later onset in adulthood. The age of onsets has two points of maximum at age 2 and around age 40.[15] New findings propose that an earlier onset leads to a longer disease duration without loss of ambulation or the need for the use of a wheelchair.[15] This was also described earlier, that later onset forms evolve more rapidly.[13]
Cause
HSP is a group of genetic disorders. It follows general inheritance rules and can be inherited in an autosomal dominant, autosomal recessive or X-linked recessive manner. The mode of inheritance involved has a direct impact on the chances of inheriting the disorder. Over 70 genotypes had been described, and over 50 genetic loci have been linked to this condition.[16] Ten genes have been identified with autosomal dominant inheritance. One of these, SPG4, accounts for ~50% of all genetically solved cases, or approximately 25% of all HSP cases.[15] Twelve genes are known to be inherited in an autosomal recessive fashion. Collectively this latter group account for ~1/3 cases.
Most altered genes have known function, but for some the function haven't been identified yet. All of them are listed in the gene list below, including their mode of inheritance. Some examples are spastin (SPG4) and paraplegin (SPG7) are both AAA ATPases.[17]
Genotypes
The genes are designated SPG (Spastic gait gene). The gene locations are in the format: chromosome - arm (short or p: long or q) - band number. These designations are for the human genes only. The locations may (and probably will) vary in other organisms. Despite the number of genes known to be involved in this condition ~40% of cases have yet to have their cause identified.[18] In the table below SPG? is used to indicate a gene that has been associated with HSP but has not yet received an official HSP gene designation.
| Genotype | OMIM | Gene symbol | Gene locus | Inheritance | Age of onset | Other names and characteristics |
|---|---|---|---|---|---|---|
| SPG1 | 303350 | L1CAM | Xq28 | X-linked recessive | Early | MASA syndrome |
| SPG2 | 312920 | PLP1 | Xq22.2 | X-linked recessive | Variable | Pelizaeus–Merzbacher disease |
| SPG3A | 182600 | ATL1 | 14q22.1 | Autosomal dominant | Early | Strumpell disease (this Wiki) |
| SPG4 | 182601 | SPAST | 2p22.3 | Autosomal dominant | Variable | |
| SPG5A | 270800 | CYP7B1 | 8q12.3 | Autosomal recessive | Variable | |
| SPG6 | 600363 | NIPA1 | 15q11.2 | Autosomal dominant | Variable | |
| SPG7 | 607259 | SPG7 | 16q24.3 | Autosomal recessive | Variable | |
| SPG8 | 603563 | KIAA0196 | 8q24.13 | Autosomal dominant | Adult | |
| SPG9A | 601162 | ALDH18A1 | 10q24.1 | Autosomal dominant | Teenage | Cataracts with motor neuronopathy, short stature and skeletal abnormalities |
| SPG9B | 616586 | ALDH18A1 | 10q24.1 | Autosomal recessive | Early | |
| SPG10 | 604187 | KIF5A | 12q13.3 | Autosomal dominant | Early | |
| SPG11 | 604360 | SPG11 | 15q21.1 | Autosomal recessive | Variable | |
| SPG12 | 604805 | RTN2 | 19q13.32 | Autosomal dominant | Early | |
| SPG13 | 605280 | HSP60 | 2q33.1 | Autosomal dominant | Variable | |
| SPG14 | 605229 | ? | 3q27–q28 | Autosomal recessive | Adult | |
| SPG15 | 270700 | ZFYVE26 | 14q24.1 | Autosomal recessive | Early | |
| SPG16 | 300266 | ? | Xq11.2 | X-linked recessive | Early | |
| SPG17 | 270685 | BSCL2 | 11q12.3 | Autosomal dominant | Teenage | |
| SPG18 | 611225 | ERLIN2 | 8p11.23 | Autosomal recessive | Early | |
| SPG19 | 607152 | ? | 9q | Autosomal dominant | Adult onset | |
| SPG20 | 275900 | SPG20 | 13q13.3 | Autosomal recessive | Early onset | Troyer syndrome |
| SPG21 | 248900 | ACP33 | 15q22.31 | Autosomal recessive | Early onset | MAST syndrome |
| SPG22 | 300523 | SLC16A2 | Xq13.2 | X-linked recessive | Early onset | Allan–Herndon–Dudley syndrome |
| SPG23 | 270750 | RIPK5 | 1q32.1 | Autosomal recessive | Early onset | Lison syndrome |
| SPG24 | 607584 | ? | 13q14 | Autosomal recessive | Early onset | |
| SPG25 | 608220 | ? | 6q23–q24.1 | Autosomal recessive | Adult | |
| SPG26 | 609195 | B4GALNT1 | 12q13.3 | Autosomal recessive | Early onset | |
| SPG27 | 609041 | ? | 10q22.1–q24.1 | Autosomal recessive | Variable | |
| SPG28 | 609340 | DDHD1 | 14q22.1 | Autosomal recessive | Early onset | |
| SPG29 | 609727 | ? | 1p31.1–p21.1 | Autosomal dominant | Teenage | |
| SPG30 | 610357 | KIF1A | 2q37.3 | Autosomal recessive | Teenage | |
| SPG31 | 610250 | REEP1 | 2p11.2 | Autosomal dominant | Early onset | |
| SPG32 | 611252 | ? | 14q12–q21 | Autosomal recessive | Childhood | |
| SPG33 | 610244 | ZFYVE27 | 10q24.2 | Autosomal dominant | Adult | |
| SPG34 | 300750 | ? | Xq24–q25 | X-linked recessive | Teenage/Adult | |
| SPG35 | 612319 | FA2H | 16q23.1 | Autosomal recessive | Childhood | |
| SPG36 | 613096 | ? | 12q23–q24 | Autosomal dominant | Teenage/Adult | |
| SPG37 | 611945 | ? | 8p21.1–q13.3 | Autosomal dominant | Variable | |
| SPG38 | 612335 | ? | 4p16–p15 | Autosomal dominant | Teenage/Adult | |
| SPG39 | 612020 | PNPLA6 | 19p13.2 | Autosomal recessive | Childhood | |
| SPG41 | 613364 | ? | 11p14.1–p11.2 | Autosomal dominant | Adolescence | |
| SPG42 | 612539 | SLC33A1 | 3q25.31 | Autosomal dominant | Variable | |
| SPG43 | 615043 | C19orf12 | 19q12 | Autosomal recessive | Childhood | |
| SPG44 | 613206 | GJC2 | 1q42.13 | Autosomal recessive | Childhood/teenage | |
| SPG45 | 613162 | NT5C2 | 10q24.32–q24.33 | Autosomal recessive | Infancy | |
| SPG46 | 614409 | GBA2 | 9p13.3 | Autosomal recessive | Variable | |
| SPG47 | 614066 | AP4B1 | 1p13.2 | Autosomal recessive | Childhood | |
| SPG48 | 613647 | AP5Z1 | 7p22.1 | Autosomal recessive | 6th decade | |
| SPG49 | 615041 | TECPR2 | 14q32.31 | Autosomal recessive | Infancy | |
| SPG50 | 612936 | AP4M1 | 7q22.1 | Autosomal recessive | Infancy | |
| SPG51 | 613744 | AP4E1 | 15q21.2 | Autosomal recessive | Infancy | |
| SPG52 | 614067 | AP4S1 | 14q12 | Autosomal recessive | Infancy | |
| SPG53 | 614898 | VPS37A | 8p22 | Autosomal recessive | Childhood | |
| SPG54 | 615033 | DDHD2 | 8p11.23 | Autosomal recessive | Childhood | |
| SPG55 | 615035 | C12orf65 | 12q24.31 | Autosomal recessive | Childhood | |
| SPG56 | 615030 | CYP2U1 | 4q25 | Autosomal recessive | Childhood | |
| SPG57 | 615658 | TFG | 3q12.2 | Autosomal recessive | Early | |
| SPG58 | 611302 | KIF1C | 17p13.2 | Autosomal recessive | Within first two decades | Spastic ataxia 2 |
| SPG59 | 603158 | USP8 | 15q21.2 | ?Autosomal recessive | Childhood | |
| SPG60 | 612167 | WDR48 | 3p22.2 | ?Autosomal recessive | Infancy | |
| SPG61 | 615685 | ARL6IP1 | 16p12.3 | Autosomal recessive | Infancy | |
| SPG62 | 615681 | ERLIN1 | 10q24.31 | Autosomal recessive | Childhood | |
| SPG63 | 615686 | AMPD2 | 1p13.3 | Autosomal recessive | Infancy | |
| SPG64 | 615683 | ENTPD1 | 10q24.1 | Autosomal recessive | Childhood | |
| SPG66 | 610009 | ARSI | 5q32 | ?Autosomal dominant | Infancy | |
| SPG67 | 615802 | PGAP1 | 2q33.1 | Autosomal recessive | Infancy | |
| SPG68 | 609541 | KLC2 | 11q13.1 | Autosomal recessive | Childhood | SPOAN syndrome |
| SPG69 | 609275 | RAB3GAP2 | 1q41 | Autosomal recessive | Infancy | Martsolf syndrome, Warburg Micro syndrome |
| SPG70 | 156560 | MARS | 12q13 | ?Autosomal dominant | Infancy | |
| SPG71 | 615635 | ZFR | 5p13.3 | ?Autosomal recessive | Childhood | |
| SPG72 | 615625 | REEP2 | 5q31 | Autosomal recessive; autosomal dominant |
Infancy | |
| SPG73 | 616282 | CPT1C | 19q13.33 | Autosomal dominant | Adult | |
| SPG74 | 616451 | IBA57 | 1q42.13 | Autosomal recessive | Childhood | |
| SPG75 | 616680 | MAG | 19q13.12 | Autosomal recessive | Childhood | |
| SPG76 | 616907 | CAPN1 | 11q13 | Autosomal recessive | Adult | |
| SPG77 | 617046 | FARS2 | 6p25 | Autosomal recessive | Childhood | |
| SPG78 | 617225 | ATP13A2 | 1p36 | Autosomal recessive | Adult | Kufor–Rakeb syndrome |
| SPG79 | 615491 | UCHL1 | 4p13 | Autosomal recessive | Childhood | |
| HSNSP | 256840 | CCT5 | 5p15.2 | Autosomal recessive | Childhood | Hereditary sensory neuropathy with spastic paraplegia |
| SPG? | SERAC1 | 6q25.3 | Juvenile | MEGDEL syndrome | ||
| SPG? | 605739 | KY | 3q22.2 | Autosomal recessive | Infancy | |
| SPG? | PLA2G6 | 22q13.1 | Autosomal recessive | Childhood | ||
| SPG? | ATAD3A | 1p36.33 | Autosomal dominant | Childhood | Harel-Yoon syndrome | |
| SPG? | KCNA2 | 1p13.3 | Autosomal dominant | Childhood | ||
| SPG? | Granulin | 17q21.31 | ||||
| SPG? | POLR3A | 10q22.3 | Autosomal recessive | |||
Pathophysiology
The major feature of HSP is a length-dependent axonal degeneration.[19] These include the crossed and uncrossed corticospinal tracts to the legs and fasciculus gracilis. The spinocerebellar tract is involved to a lesser extent. Neuronal cell bodies of degenerating axons are preserved and there is no evidence of primary demyelination.[16] Loss of anterior horn cells of the spinal cord are observed in some cases. Dorsal root ganglia, posterior roots and peripheral nerves are not directly affected.
HSP affects several pathways in motor neurons. Many genes were identified and linked to HSP. It remains a challenge to accurately define the key players in each of the affected pathways, mainly because many genes have multiple functions and are involved in more than one pathway .
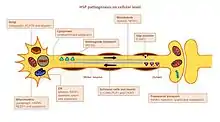
Axon pathfinding
Pathfinding is important for axon growth to the right destination (e.g. another nerve cell or a muscle). Significant for this mechanism is the L1CAM gene, a cell surface glycoprotein of the immunoglobulin superfamily. Mutations leading to a loss-of-function in L1CAM are also found in other X-linked syndromes. All of these disorders display corticospinal tract impairment (a hallmark feature of HSP). L1CAM participates in a set of interactions, binding other L1CAM molecules as well as extracellular cell adhesion molecules, integrins, and proteoglycans or intracellular proteins like ankyrins.
The pathfinding defect occurs via the association of L1CAM with neuropilin-1. Neuropilin-1 interacts with Plexin-A proteins to form the Semaphorin-3A receptor complex. Semaphorin-a3A is then released in the ventral spinal cord to steer corticospinal neurons away from the midline spinal cord / medullary junction. If L1CAM does not work correctly due to a mutation, the cortiocospinal neurons are not directed to the correct position and the impairment occurs.[3]
Lipid metabolism
Axons in the central and peripheral nervous system are coated with an insulation, the myelin layer, to increase the speed of action potential propagation. Abnormal myelination in the CNS is detected in some forms of hsp HSP.[20] Several genes were linked to myelin malformation, namely PLP1, GFC2 and FA2H.[3] The mutations alter myelin composition, thickness and integrity.
Endoplasmic reticulum (ER) is the main organelle for lipid synthesis. Mutations in genes encoding proteins that have a role in shaping ER morphology and lipid metabolism were linked to HSP. Mutations in ATL1, BSCL2 and ERLIN2 alter ER structure, specifically the tubular network and the formation of three-way junctions in ER tubules. Many mutated genes are linked to abnormal lipid metabolism. The most prevalent effect is on arachidonic acid (CYP2U1) and cholesterol (CYP7B1) metabolism, phospholipase activity (DDHD1 and DDHD2), ganglioside formation (B4GALNT-1) and the balance between carbohydrate and fat metabolism (SLV33A1).[3][21][20]
Endosomal trafficking
Neurons take in substances from their surrounding by endocytosis. Endocytic vesicles fuse to endosomes in order to release their content. There are three main compartments that have endosome trafficking: Golgi to/from endosomes; plasma membrane to/from early endosomes (via recycling endosomes) and late endosomes to lysosomes. Dysfunction of endosomal trafficking can have severe consequences in motor neurons with long axons, as reported in HSP. Mutations in AP4B1 and KIAA0415 are linked to disturbance in vesicle formation and membrane trafficking including selective uptake of proteins into vesicles. Both genes encode proteins that interact with several other proteins and disrupt the secretory and endocytic pathways.[20]
Mitochondrial function
Mitochondrial dysfunctions have been connected with developmental and degenerative neurological disorders. Only a few HSP genes encode mitochondrial proteins. Two mitochondrial resident proteins are mutated in HSP: paraplegin and chaperonin 60. Paraplegin is a m-AAA metalloprotease of the inner mitochondrial membrane. It functions in ribosomal assembly and protein quality control. The impaired chaperonin 60 activity leads to impaired mitochondrial quality control. Two genes DDHD1 and CYP2U1 have shown alteration of mitochondrial architecture in patient fibroblasts. These genes encode enzymes involved in fatty-acid metabolism.
Diagnosis
Initial diagnosis of HSPs relies upon family history, the presence or absence of additional signs and the exclusion of other nongenetic causes of spasticity, the latter being particular important in sporadic cases.[7]
Cerebral and spinal MRI is an important procedure performed in order to rule out other frequent neurological conditions, such as multiple sclerosis, but also to detect associated abnormalities such as cerebellar or corpus callosum atrophy as well as white matter abnormalities. Differential diagnosis of HSP should also exclude spastic diplegia which presents with nearly identical day-to-day effects and even is treatable with similar medicines such as baclofen and orthopedic surgery; at times, these two conditions may look and feel so similar that the only perceived difference may be HSP's hereditary nature versus the explicitly non-hereditary nature of spastic diplegia (however, unlike spastic diplegia and other forms of spastic cerebral palsy, HSP cannot be reliably treated with selective dorsal rhizotomy).
Ultimate confirmation of HSP diagnosis can only be provided by carrying out genetic tests targeted towards known genetic mutations.
Classification
Hereditary spastic paraplegias can be classified based on the symptoms; mode of inheritance; the patient's age at onset; the affected genes; and biochemical pathways involved.
Treatment
No specific treatment is known that would prevent, slow, or reverse HSP. Available therapies mainly consist of symptomatic medical management and promoting physical and emotional well-being. Therapeutics offered to HSP patients include:
- Baclofen – a voluntary muscle relaxant to relax muscles and reduce tone. This can be administered orally or intrathecally. (Studies in HSP [22][23][24])
- Tizanidine – to treat nocturnal or intermittent spasms (studies available [25][26])
- Diazepam and clonazepam – to decrease intensity of spasms
- Oxybutynin chloride – an involuntary muscle relaxant and spasmolytic agent, used to reduce spasticity of the bladder in patients with bladder control problems
- Tolterodine tartrate – an involuntary muscle relaxant and spasmolytic agent, used to reduce spasticity of the bladder in patients with bladder control problems
- Cro System – to reduce muscle overactivity (existing studies for spasticity [27][28][29])
- Botulinum toxin – to reduce muscle overactivity (existing studies for HSP patients[30][31])
- Antidepressants (such as selective serotonin re-uptake inhibitors, tricyclic antidepressants and monoamine oxidase inhibitors) – for patients experiencing clinical depression
- Physical therapy – to restore and maintain the ability to move; to reduce muscle tone; to maintain or improve range of motion and mobility; to increase strength and coordination; to prevent complications, such as frozen joints, contractures, or bedsores.
Prognosis
Although HSP is a progressive condition, the prognosis for individuals with HSP varies greatly. It primarily affects the legs although there can be some upperbody involvement in some individuals. Some cases are seriously disabling whilst others leave people able to do most ordinary activities to an ordinary extent without needing adjustments. The majority of individuals with HSP have a normal life expectancy.[14]
Epidemiology
Worldwide, the prevalence of all hereditary spastic paraplegias combined is estimated to be 2 to 6 in 100,000 people.[32] A Norwegian study of more than 2.5 million people published in March 2009 has found an HSP prevalence rate of 7.4/100,000 of population – a higher rate, but in the same range as previous studies. No differences in rate relating to gender were found, and average age at onset was 24 years.[33] In the United States, Hereditary Spastic Paraplegia is listed as a "rare disease" by the Office of Rare Diseases (ORD) of the National Institutes of Health which means that the disorder affects less than 200,000 people in the US population.[32]
References
- Fink, John K. (2003-08-01). "The hereditary spastic paraplegias: nine genes and counting". Archives of Neurology. 60 (8): 1045–1049. doi:10.1001/archneur.60.8.1045. ISSN 0003-9942. PMID 12925358.
- Depienne, Christel; Stevanin, Giovanni; Brice, Alexis; Durr, Alexandra (2007-12-01). "Hereditary spastic paraplegias: an update". Current Opinion in Neurology. 20 (6): 674–680. doi:10.1097/WCO.0b013e3282f190ba. ISSN 1350-7540. PMID 17992088. S2CID 35343501.
- Blackstone, Craig (21 July 2012). "Cellular Pathways of Hereditary Spastic Paraplegia". Annual Review of Neuroscience. 35 (1): 25–47. doi:10.1146/annurev-neuro-062111-150400. PMC 5584684. PMID 22540978.
- De Matteis, Maria Antonietta; Luini, Alberto (2011-09-08). "Mendelian disorders of membrane trafficking". The New England Journal of Medicine. 365 (10): 927–938. doi:10.1056/NEJMra0910494. ISSN 1533-4406. PMID 21899453. S2CID 14772080.
- Faber I, Pereira ER, Martinez AR, França M Jr, Teive HA (November 2013). "Hereditary spastic paraplegia from 1880 to 2017: an historical review". Arquivos de Neuro-Psiquiatria. Brazilian Academy of Neurology. 75 (11): 813–818. doi:10.1590/0004-282X20170160. PMID 29236826.
- Lorrain, Maurice. Contribution à l'étude de la paraplégie spasmodique familiale: travail de la clinique des maladies du système nerveux à la Salpêtrière. G. Steinheil, 1898.
- Harding, AE (1983). "Classification of the hereditary ataxias and paraplegias". Lancet. New York. 1 (8334): 1151–5. doi:10.1016/s0140-6736(83)92879-9. PMID 6133167. S2CID 6780732.
- McAndrew CR, Harms P (2003). "Paraesthesias during needle-through-needle combined spinal epidural versus single-shot spinal for elective caesarean section". Anaesthesia and Intensive Care. 31 (5): 514–517. doi:10.1177/0310057X0303100504. PMID 14601273.
- Fjermestad, Krister W.; Kanavin, Øivind J.; Næss, Eva E.; Hoxmark, Lise B.; Hummelvoll, Grete (2016-07-13). "Health survey of adults with hereditary spastic paraparesis compared to population study controls". Orphanet Journal of Rare Diseases. 11 (1): 98. doi:10.1186/s13023-016-0469-0. ISSN 1750-1172. PMC 4944497. PMID 27412159.
- Chaudhuri, Abhijit; Behan, Peter O. (2004-03-20). "Fatigue in neurological disorders". Lancet. 363 (9413): 978–988. doi:10.1016/S0140-6736(04)15794-2. ISSN 1474-547X. PMID 15043967. S2CID 40500803.
- "Hereditary spastic paraplegia". nhs.uk. 2017-10-18. Retrieved 2018-01-28.
- Fink JK (2003). "The Hereditary Spastic Paraplegias". Archives of Neurology. 60 (8): 1045–1049. doi:10.1001/archneur.60.8.1045. PMID 12925358.
- Harding AE (1981). "Hereditary "pure" spastic paraplegia: a clinical and genetic study of 22 families". Journal of Neurology, Neurosurgery, and Psychiatry. 44 (10): 871–883. doi:10.1136/jnnp.44.10.871. PMC 491171. PMID 7310405.
- Depienne C, Stevanin G, Brice A, Durr A (2007). "Hereditary Spastic Paraplegia: An Update". Current Opinion in Neurology. 20 (6): 674–680. doi:10.1097/WCO.0b013e3282f190ba. PMID 17992088. S2CID 35343501.
- Schüle, Rebecca; Wiethoff, Sarah; Martus, Peter; Karle, Kathrin N.; Otto, Susanne; Klebe, Stephan; Klimpe, Sven; Gallenmüller, Constanze; Kurzwelly, Delia (2016-04-01). "Hereditary spastic paraplegia: Clinicogenetic lessons from 608 patients". Annals of Neurology. 79 (4): 646–658. doi:10.1002/ana.24611. ISSN 1531-8249. PMID 26856398. S2CID 10558032.
- Schüle R, Schöls L (2011) Genetics of hereditary spastic paraplegias. Semin Neurol 31(5):484-493
- Wang YG, Shen L (2009) AAA ATPases and hereditary spastic paraplegia. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 26(3):298-301
- Helbig KL, Hedrich UB, Shinde DN, Krey I, Teichmann AC, Hentschel J, Schubert J, Chamberlin AC, Huether R, Lu HM4, Alcaraz WA, Tang S, Jungbluth C, Dugan SL, Vainionpää L, Karle KN, Synofzik M, Schöls L, Schüle R, Lehesjoki AE, Helbig I, Lerche H, Lemke JR (2016) A recurrent mutation in KCNA2 as a novel cause of hereditary spastic paraplegia and ataxia. Ann Neurol 80(4)
- Wharton SB, McDermott CJ, Grierson AJ, Wood JD, Gelsthorpe C, Ince PG, Shaw PJ (2003) The cellular and molecular pathology of the motor system in hereditary spastic paraparesis due to mutation of the spastin gene. J Neuropathol Exp Neurol 62:1166–1177
- Noreau, A., Dion, P.A. & Rouleau, G.A., 2014. Molecular aspects of hereditary spastic paraplegia. Experimental Cell Research, 325(1), pp.18–26
- Lo Giudice, T. et al., 2014. Hereditary spastic paraplegia: Clinical-genetic characteristics and evolving molecular mechanisms. Experimental Neurology, 261, pp.518–539.
- Margetis K, Korfias S, Boutos N, Gatzonis S, Themistocleous M, Siatouni A, et al. Intrathecal baclofen therapy for the symptomatic treatment of hereditary spastic paraplegia. Clinical Neurology and Neurosurgery. 2014;123:142-5.
- Heetla HW, Halbertsma JP, Dekker R, Staal MJ, van Laar T. Improved Gait Performance in a Patient With Hereditary Spastic Paraplegia After a Continuous Intrathecal Baclofen Test Infusion and Subsequent Pump Implantation: A Case Report. Archives of Physical Medicine and Rehabilitation. 2015;96(6):1166-9.
- Klebe S, Stolze H, Kopper F, Lorenz D, Wenzelburger R, Deuschl G, et al. Objective assessment of gait after intrathecal baclofen in hereditary spastic paraplegia. Journal of Neurology. 2005;252(8):991-3.
- Knutsson E, Mårtensson A, Gransberg L. Antiparetic and antispastic effects induced by tizanidine in patients with spastic paresis. Journal of the Neurological Sciences. 1982;53(2):187-204.
- Bes A, Eyssette M, Pierrot-Deseilligny E, Rohmer F, Warter JM. A multi-centre, double-blind trial of tizanidine, a new antispastic agent, in spasticity associated with hemiplegia. Current Medical Research and Opinion. 1988;10(10):709-18.
- Celletti C, Camerota F. Preliminary evidence of focal muscle vibration effects on spasticity due to cerebral palsy in a small sample of Italian children. Clin Ter. 162(5): 125–8. 2011
- Caliandro P, Celletti C, Padua L, Minciotti I, Russo G, Granata G, La Torre G, Granieri E, Camerota F. Focal muscle vibration in the treatment of upper limb spasticity: a pilot randomized controlled trial in patients with chronic stroke. Arch Phys Med Rehabil. 93(9):1656-61. 2012.
- . Casale R1, Damiani C, Maestri R, Fundarò C, Chimento P, Foti C. Focalized 100 Hz vibration improves function and reduces upper limb spasticity: a double-blind controlled study. Eur J Phys Rehabil Med. 2014 Oct;50(5):495-504. 2014.
- Hecht MJ, Stolze H, Auf Dem Brinke M, Giess R, Treig T, Winterholler M, et al. Botulinum neurotoxin type A injections reduce spasticity in mild to moderate hereditary spastic paraplegia— Report of 19 cases. Movement Disorders. 2008;23(2):228-33.
- de Niet M, de Bot ST, van de Warrenburg BP, Weerdesteyn V, Geurts AC. Functional effects of botulinum toxin type-A treatment and subsequent stretching of spastic calf muscles: A study in patients with hereditary spastic paraplegia. Journal of rehabilitation medicine. 2015;47(2):147-53.
- National Institute of Health (2008). "Hereditary Spastic Paraplegia Information Page". Archived from the original on 2014-02-21. Retrieved 2008-04-30.
- Erichsen, AK; Koht, J; Stray-Pedersen, A; Abdelnoor, M; Tallaksen, CM (June 2009). "Prevalence of hereditary ataxia and spastic paraplegia in southeast Norway: a population-based study". Brain. 132 (Pt 6): 1577–88. doi:10.1093/brain/awp056. PMID 19339254.
Further reading
- GeneReviews/NCBI/NIH/UW entry on Spastic Paraplegia 3A
- GeneReviews/NCBI/NIH/UW entry on Hereditary Spastic Paraplegia Overview
- Warner, Tom (January–February 2007). "Hereditary Spastic Paraplegia" (PDF). Advances in Clinical Neuroscience and Rehabilitation. 6 (6): 16–17. Archived from the original (PDF) on 2020-11-27. Retrieved 2013-05-20.