Genodermatosis
Genodermatosis is a hereditary skin disease with three inherited modes including single gene inheritance, multiple gene inheritance and chromosome inheritance.[1] There are many different types of genodermatosis, the prevalence of genodermatosis ranges from 1 per 6000 people to 1 per 500,000 people.[2] Genodermatosis has influence on the texture, color and structure of skin cuticle and connective tissue, specific lesion site and clinical manifestations on the body vary depending on the type.[3] In the spite of the variety and complexity of genodermatosis, there are still some common methods that can help people diagnose.[4] After diagnosis, different types of genodermatosis require different levels of therapy including interventions, nursing interventions and treatments.[5] Among that, research of therapy for some new, complex and rare types are still in the developing stage.[6] The impact of genodermatosis not only can be seen in body but also can be seen in all aspects of patients' life, including but not limited to psychological, family life, economic conditions and social activities.[5][7] Accordingly, the patients need treatment, support and help in these areas.[5]
| Genodermatosis | |
|---|---|
| Other names | genodermatoses |
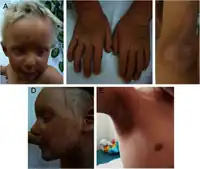 | |
| A patient with Clouston's hidrotic ectodermal dysplasia, one of the rare genodermatosis. | |
| Specialty | Dermatology, Medical genetics |
| Causes | Family history; gene mutation; chromosome abnormality |
Hereditary modes
Genodermatosis is inherited in three modes: single gene inheritance, multiple gene inheritance and chromosome inheritance.[1]
Single gene (monogenic)
Single-gene inheritance of genodermatosis refers to the inheritance of a skin disease caused by one genetic abnormality and single gene heredity is divided into four kinds mainly.[2]
Autosomal dominant inheritance
The first kind is autosomal dominant inheritance, in this kind of inheritance, patients can be of any sex and their genodermatosis are often inherited from one of the parents.[2] Cases of skin disease that may be inherited in this kind of mode include epidermolysis bullosa simplex (EBS), acute intermittent porphyria, white sponge nevus, ichthyosis, epidermolytic palmoplantar keratoderma, hereditary benign intraepithelial dyskeratosis and so on.[1][2][8][9]
Autosomal recessive inheritance
The second kind is autosomal recessive inheritance, in this kind of inheritance, patients can be of any sex and inbreeding tends to lead to this inheritance.[2] Cases of skin disease that may be inherited in this kind of mode include epidermolysis bullosa, xeroderma pigmentosum, acrodermatitis enteropathica, ichthyosis and so on.[1][2]
X-linked dominant inheritance
The third kind is X-linked dominant inheritance, in this kind of inheritance, patients can be of any sex.[2] Male patients can pass the disease on to their sons and the chances of female patients passing it to their daughters or sons are almost equal.[2] Cases of skin disease that may be inherited in this kind of mode include incontinentia pigmenti, focal dermal hypoplasia and so on.[1][8]
X-linked recessive inheritance
The last kind is X-linked recessive inheritance, in this kind of inheritance, patients can be of any sex and the prevalence in men is higher than that in women.[2] Male patients cannot pass the disease on to their sons.[2] Cases of skin disease that may be inherited in this kind of mode include fabry disease, anhidrotic ectodermal dysplasia, dyskeratosis congenita and so on.[1][2]
Multiple gene (polygenic)
Multiple-gene inheritance of genodermatosis refers to the inheritance of a skin disease caused by multiple genetic abnormalities.[2] Cases of skin disease that may be inherited in this mode include vitiligo, psoriasis, pemphigus vulgaris, systemic lupus erythematosus and so on.[2][8]
Chromosome
Chromosome inheritance of genodermatosis refers to the inheritance of a skin disease caused by chromosome abnormality.
The same disease can be inherited in different modes. For instance, epidermolysis bullosa can be inherited in the mode of autosomal dominant or in the mode of autosomal recessive.
Types
.jpg.webp)
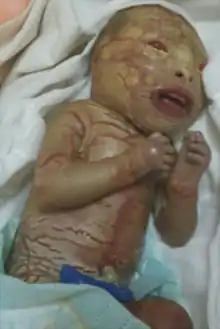

Genodermatosis has many types, many of which are rare.
Ichthyosis
Ichthyosis refers mainly to ichthyosis vulgaris, a common genodermatosis, people with this disease have a fishy, dry skin, which usually appears in early childhood and may disappear in adulthood.[10] The prevalence of ichthyosis vulgaris is high, affecting almost 1 per 250 people.[11] There are also rare types of ichthyosis, such as epidermolytic hyperkeratosis, harlequin ichthyosis and so on.[8]
Michelin tyre baby syndrome
Michelin tyre baby syndrome is a rare genodermatosis that occurs at birth, the skin of the patients is stacked symmetrically in layers like the image of the Michelin tyre's mascot, which is also how this disease got its name.[12]
Epidermolysis bullosa
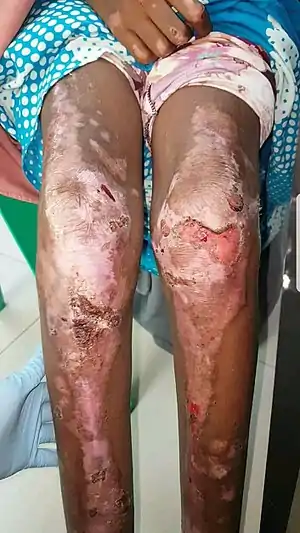
Epidermolysis bullosa is a rare type of genodermatosis, people with this disease have blisters on their skin and this disease is never completely cured for a lifetime.[13] Epidermolysis bullosa is mainly subdivided into four types: dystrophic epidermolysis bullosa, epidermolysis bullosa simplex, junctional epidermolysis bullosa and kindler syndrome.[13] Almost 1 in 50,000 people has epidermolysis bullosa.[14]
Pachyonychia congenita

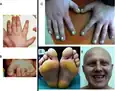
Pachyonychia congenita is a rare type of genodermatosis, its clinical manifestations are abnormal enlargement of fingernails or toenails, excessive or poor palmoplantar keratinization, excessive sweating in the palmar or the plantar.[15] Between 5000 and 10000 people in the world have pachyonychia congenita.[16]
Epidermolytic palmoplantar keratoderma
Epidermolytic palmoplantar keratoderma often appears at birth and it is almost impossible to be cured completely.[9] The clinical manifestations of this disease include excessive palmoplantar keratinization, the palmar or plantar become yellow divergently with around the edges or abnormally excessive sweating and clinical manifestation appear in a symmetrical form on the body.[9]
Hereditary benign intraepithelial dyskeratosis
Hereditary benign intraepithelial dyskeratosis is a rare type of genodermatosis that may occur in infancy and early childhood, its symptoms often appear in the patients' eyes and mouths.[17] The patients' eyes appear red due to the dilatation of superficial vessels and appearance of conjunctival plaques in their eyes, patients may have variable-size, thick, soft and white plaques in their mouth.[17] Spring is an acute episode of symptoms, such as itching, erythema, photophobia and so on.[17]
Epidermolytic hyperkeratosis
Epidermolytic hyperkeratosis is a rare genodermatosis which is also referred to as disorder of cornification type 3 and bullous congenital ichthyosiform erthroderma, affecting almost 1 per 200,000 - 300,000 people.[18] They also stated that its clinical manifestations often begin at birth with large rashes all over the body, and the patients' skin will be so sensitive that even mild wounds can cause blisters and peeling. Potential complications of this disease include electrolyte disturbances, sepsis and so on.[18]
Hidrotic ectodermal dysplasia
Hidrotic ectodermal dysplasia is a rare genodermatosis which is also known as Clouston syndrome. The patients' nails may be too thick, too brittle, too bent or have different colors, their hair also appear mottled, sparse and other abnormalities.[19] These symptoms often begin when the patient is an infant.[19]
Diagnostic methods
Genodermatosis is often rare and varied, but diagnostic methods have some commonalities, the diagnosis of rare genodermatosis is basically divided into six steps: 1. Each genodermatosis has different clinical manifestations. People can observe the special features and changes of the skin to judge whether they are sick or not. Features will be different in different age groups, so it is necessary to observe and record the special features of the skin in each age group; 2. Genodermatosis is a hereditary disease, knowing as much as possible about a detailed and complete family history helps in screening and diagnosis; 3. People can do a detailed physical examination to observe the special features and manifestations of other organs besides skin. It can help to narrow down the disease and make a definitive diagnosis; 4. People can carry out laboratory tests such as skin biopsies with high-tech and precise scientific instruments to have further results; 5. Different genodermatosis and their clinical manifestations may be caused by abnormalities in the same gene, and abnormalities in different genes may also lead to the same clinical manifestations. In order to have a definitive diagnosis or identify complex and specific types of genodermatosis, genotype–phenotype correlation needs attention; 6. If the above five steps fails to help people who suspected of having a genodermatosis to obtain the diagnosis result, they should keep all their information such as the diagnosis record and the clinical manifestations at different stages, and continue to record the changes of the body, waiting with a positive attitude, the future medicine may give the answer.[4]
Therapy
The therapy of genodermatosis not only needs to take care of patients' skin, reduce their pain and prevent complications, but also needs to carry out mental support for patients and their families.[5]
Prevention and care
Different types of genodermatosis require different kinds of prevention and care.
Ichthyosis
There is no radical therapeutic method for ichthyosis, but some care can ease the symptoms.[10] To prevent skin thickening and hydrate skin, patients can apply a cream containing alpha hydroxy acids and patients can also be treated with antibiotics for subsequent infections.[10]
Epidermolysis bullosa
For epidermolysis bullosa, daily care is important. When treating a wound, keep it clean and reduce friction, the bandages and dressing used must be non-sticky and gentle, and the patient should wear loose clothing to avoid damaging the wound.[13][14]
Epidermolytic hyperkeratosis
The treatment of Epidermolytic hyperkeratosis is mainly control and alleviate symptoms, and good nursing can reduce the incidence of complications like electrolyte disturbances and sepsis.[18] To improve the look and feeling of the skin, patients can apply a cream containing alpha hydroxy acids, glycerol and urea, and if necessary, patients can use antibiotics to control secondary infections.[18]
Pachyonychia congenita
There is no radical therapeutic method for pachyonychia congenita, but some care can ease the symptoms.[16] Patients can polish and trim thickened nails, some of them can use retinoids to relieve symptoms but it may increase pain.[16]
Neurofibromatosis type I
Selumetinib and trametinib have been shown to reduce and control tumor growth in people with neurofibromatosis type I, reducing the likelihood of malignant lesions.[20]
Therapeutic methods
There are some new and developing genetic therapies available in genodermatosis.[20]
In the case of X-linked hypohidrotic ectodermal dysplasia, unborn babies diagnosed with this disease can be treated in their mothers' womb. Providing regulatory proteins in the womb during a critical period of infant growth may help correct the development of babies' sweat glands.[20]
Ustekinumab is a biologic therapy that can be used in a variety of genodermatosis such as congenital ichthyosis, psoriasis, deficiency of interleukin-36 receptor antagonist (DITRA) and so on.[20]
A new topical method could treat skin abnormalities in rare inherited lipid metabolic diseases.[21] This method obstructs abnormal mevalonate by topical application of lovastatin so that the production and cumulation of poisonous metabolic intermediates can be inhibited as much as possible and takes the place of the lacking lipid in the skin by topical application of cholesterol.[20][21] The idea that similar treatments could be developed for other genodermatosis was also pointed out at the annual conference of the European Society of Dermatology and Venereology.[21]
For epidermolysis bullosa, a method called CRISPR can be used to treat this disease by gene analysis, modification and substitution, but the method is ethically controversial because it consents to editing genes highly.[20]
Treatments for some rare diseases are still being studied. The therapy of genodermatosis requires the updating of technology, and the development of technology depends on the continuous understanding of the mechanism of the disease. Research on the treatment of genodermatosis is at a positive stage of development.[20]
Effects
Genodermatosis affects patients in many ways. Genodermatosis is a kind of skin disease, it affects the texture, color and structure of the cuticle and connective tissue of the skin, some of which can cause abnormalities in other organs.[3] On the social side, because the genodermatosis makes the patients' skin and appearance different from the ordinary people and makes them have limitations in some activities, they more or less encounter obstacles in the process of making friends, seeking a mate, going to school and entering the workplace.[5][7] Difficulties in communicating with others as well as worldly prejudice may affect their mental health. Patients are also affected by genodermatosis in terms of family life.[5][7] Because of the behavioral disorders and treatment of certain genodermatosis, families need to spend more time caring for the patient, and the patient may have more concerns and considerations about procreating children due to the disease. In terms of economy, the treatment of genodermatosis is not a simple and short process, which will generate additional family expenses and increase economic pressure on patients and their families.[7]
See also
References
- Babu, N. Aravindha; Rajesh, E.; Krupaa, Jayasri; Gnananandar, G. (2015). "Genodermatoses". Journal of Pharmacy & Bioallied Sciences. 7 (Suppl 1): S203–S206. doi:10.4103/0975-7406.155903. PMC 4439671. PMID 26015711.
- Fields, D. (2019, June). Types of Genodermatoses. Retrieved September 08, 2020, from https://www.news-medical.net/health/Types-of-Genodermatoses.aspx
- Bayart, Cheryl B.; Brandling-Bennett, Heather A. (2018). "Congenital and Hereditary Disorders of the Skin". Avery's Diseases of the Newborn. pp. 1475–1494.e1. doi:10.1016/B978-0-323-40139-5.00104-2. ISBN 978-0-323-40139-5.
- Tantcheva-Poór, Iliana; Oji, Vinzenz; Has, Cristina (October 2016). "A multistep approach to the diagnosis of rare genodermatoses". Journal der Deutschen Dermatologischen Gesellschaft. 14 (10): 969–986. doi:10.1111/ddg.13140. PMID 27767270. S2CID 46823821.
- Fondation René Touraine. (n.d.). Genodermatoses & Rare Skin Disorders - a public health priority. Retrieved September 08, 2020, from https://www.frt-rareskin.org/Genodermatoses-Rare-Skin-Disorders
- Chamcheu, Jean Christopher; Wood, Gary S.; Siddiqui, Imtiaz A.; Syed, Deeba N.; Adhami, Vaqar M.; Teng, Joyce M.; Mukhtar, Hasan (July 2012). "Progress towards genetic and pharmacological therapies for keratin genodermatoses: current perspective and future promise". Experimental Dermatology. 21 (7): 481–489. doi:10.1111/j.1600-0625.2012.01534.x. PMC 3556927. PMID 22716242.
- Dufresne, H.; Hadj-Rabia, S.; Bodemer, C. (November 2018). "Impact of a rare chronic genodermatosis on family daily life: the example of epidermolysis bullosa". British Journal of Dermatology. 179 (5): 1177–1178. doi:10.1111/bjd.16710. PMID 29710387. S2CID 207077766.
- DeStefano, G. M.; Christiano, A. M. (1 October 2014). "The Genetics of Human Skin Disease". Cold Spring Harbor Perspectives in Medicine. 4 (10): a015172. doi:10.1101/cshperspect.a015172. PMC 4200211. PMID 25274756.
- Chen, Nan; Sun, Jingying; Song, Yali; Wei, Xinjing; Shi, Yan; Zhang, Li (September 2017). "A novel mutation of KRT9 gene in a Chinese Han pedigree with epidermolytic palmoplantar keratoderma". Journal of Cosmetic Dermatology. 16 (3): 402–406. doi:10.1111/jocd.12263. PMID 27726289. S2CID 23426593.
- National Center for Advancing Translational Sciences. (2013). Ichthyosis vulgaris. Retrieved from https://rarediseases.info.nih.gov/diseases/6752/ichthyosis-vulgaris
- Foundation for Ichthyosis & Related Skin Types. (n.d.). Press Release: Ichthyosis Incidence Study: Foundation for Ichthyosis & Related Skin Types (FIRST). Retrieved from http://www.firstskinfoundation.org/new-study-estimates-incidence-of-moderate-to-severe-ichthyosis
- Dhingra, Dhulika; Sethi, G. R.; Mantan, Mukta (August 2013). "Michelin tyre baby: A rare genodermatosis". Indian Pediatrics. 50 (8): 808. doi:10.1007/s13312-013-0208-8. PMID 24036654. S2CID 38318738.
- Queensland Government. (2017). Epidermolysis bullosa fact sheet. Retrieved October 11, 2020, from https://www.childrens.health.qld.gov.au/fact-sheet-epidermolysis-bullosa/
- National Organization for Rare Disorders (NORD). (2019).Epidermolysis Bullosa. Retrieved from https://rarediseases.org/rare-diseases/epidermolysis-bullosa/
- Agarwal, Puneet; Chhaperwal, MahendraK; Singh, Apurva; Verma, Arvind; Nijhawan, Manisha; Singh, Kishore; Mathur, Dinesh (2013). "Pachyonychia congenita: A rare genodermatosis". Indian Dermatology Online Journal. 4 (3): 225–7. doi:10.4103/2229-5178.115527. PMC 3752484. PMID 23984242.
- National Organization for Rare Disorders (NORD). (2018). Pachyonychia Congenita. Retrieved from
- O'Neill, M. J. (2013, May 17). DYSKERATOSIS, HEREDITARY BENIGN INTRAEPITHELIAL; HBID. Retrieved from
- Kwak, Juliann; Maverakis, Emanual (8 September 2006). "Epidermolytic hyperkeratosis". Dermatology Online Journal. 12 (5): 6. doi:10.5070/D38HV5R331. PMID 16962021. ProQuest 68849936.
- National Center for Advancing Translational Sciences. (2020). Clouston syndrome. Retrieved from
- Silverberg, Nanette (July 2020). "Emerging therapies in genodermatoses". Clinics in Dermatology. 38 (4): 462–466. doi:10.1016/j.clindermatol.2020.03.006. PMID 32972604. S2CID 216334904.
- Jancin, Bruce (16 November 2011). "Topical Treatment for Genodermatosis Called 'Genius'". Dermatology News.