Protein tertiary structure
Protein tertiary structure is the three dimensional shape of a protein. The tertiary structure will have a single polypeptide chain "backbone" with one or more protein secondary structures, the protein domains. Amino acid side chains may interact and bond in a number of ways. The interactions and bonds of side chains within a particular protein determine its tertiary structure. The protein tertiary structure is defined by its atomic coordinates. These coordinates may refer either to a protein domain or to the entire tertiary structure.[1][2] A number of tertiary structures may fold into a quaternary structure.[3]
-en.svg.png.webp)
-en.svg){kind=link}
History
The science of the tertiary structure of proteins has progressed from one of hypothesis to one of detailed definition. Although Emil Fischer had suggested proteins were made of polypeptide chains and amino acid side chains, it was Dorothy Maud Wrinch who incorporated geometry into the prediction of protein structures. Wrinch demonstrated this with the Cyclol model, the first prediction of the structure of a globular protein.[4] Contemporary methods are able to determine, without prediction, tertiary structures to within 5 Å (0.5 nm) for small proteins (<120 residues) and, under favorable conditions, confident secondary structure predictions.
Determinants
Thermostability
A protein folded into its native state or native conformation typically has a lower Gibbs free energy (a combination of enthalpy and entropy) than the unfolded conformation. A protein will tend towards low-energy conformations, which will determine the protein's fold in the cellular environment. Because many similar conformations will have similar energies, protein structures are dynamic, fluctuating between these similar structures.
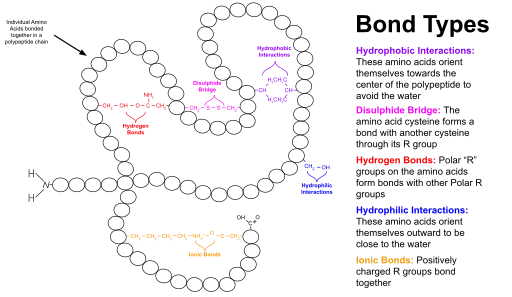
Globular proteins have a core of hydrophobic amino acid residues and a surface region of water-exposed, charged, hydrophilic residues. This arrangement may stabilize interactions within the tertiary structure. For example, in secreted proteins, which are not bathed in cytoplasm, disulfide bonds between cysteine residues help to maintain the tertiary structure. There is a commonality of stable tertiary structures seen in proteins of diverse function and diverse evolution. For example, the TIM barrel, named for the enzyme triosephosphateisomerase, is a common tertiary structure as is the highly stable, dimeric, coiled coil structure. Hence, proteins may be classified by the structures they hold. Databases of proteins which use such a classification include SCOP and CATH.
Kinetic traps
Folding kinetics may trap a protein in a high-energy conformation, i.e. a high-energy intermediate conformation blocks access to the lowest-energy conformation. The high-energy conformation may contribute to the function of the protein. For example, the influenza hemagglutinin protein is a single polypeptide chain which when activated, is proteolytically cleaved to form two polypeptide chains. The two chains are held in a high-energy conformation. When the local pH drops, the protein undergoes an energetically favorable conformational rearrangement that enables it to penetrate the host cell membrane.
Metastability
Some tertiary protein structures may exist in long-lived states that are not the expected most stable state. For example, many serpins (serine protease inhibitors) show this metastability. They undergo a conformational change when a loop of the protein is cut by a protease.[5][6][7]
Chaperone proteins
It is commonly assumed that the native state of a protein is also the most thermodynamically stable and that a protein will reach its native state, given its chemical kinetics, before it is translated. Protein chaperones within the cytoplasm of a cell assist a newly synthesised polypeptide to attain its native state. Some chaperone proteins are highly specific in their function, for example, protein disulfide isomerase; others are general in their function and may assist most globular proteins, for example, the prokaryotic GroEL/GroES system of proteins and the homologous eukaryotic heat shock proteins (the Hsp60/Hsp10 system).
Cytoplasmic environment
Prediction of protein tertiary structure relies on knowing the protein's primary structure and comparing the possible predicted tertiary structure with known tertiary structures in protein data banks. This only takes into account the cytoplasmic environment present at the time of protein synthesis to the extent that a similar cytoplasmic environment may also have influenced the structure of the proteins recorded in the protein data bank.
Ligand binding
The structure of a protein, such as an enzyme, may change upon binding of its natural ligands, for example a cofactor. In this case, the structure of the protein bound to the ligand is known as holo structure, while the unbound protein has an apo structure.[8]
Structure stabilized by the formation of weak bonds between amino acid side chains - Determined by the folding of the polypeptide chain on itself (nonpolar residues are located inside the protein, while polar residues are mainly located outside) - Envelopment of the protein brings the protein closer and relates a-to located in distant regions of the sequence - Acquisition of the tertiary structure leads to the formation of pockets and sites suitable for the recognition and the binding of specific molecules (biospecificity).
Determination
The knowledge of the tertiary structure of soluble globular proteins is more advanced than that of membrane proteins because the former are easier to study with available technology.
X-ray crystallography
X-ray crystallography is the most common tool used to determine protein structure. It provides high resolution of the structure but it does not give information about protein's conformational flexibility.
NMR
Protein NMR gives comparatively lower resolution of protein structure. It is limited to smaller proteins. However, it can provide information about conformational changes of a protein in solution.
Cryogenic electron microscopy
Cryogenic electron microscopy (cryo-EM) can give information about both a protein's tertiary and quaternary structure. It is particularly well-suited to large proteins and symmetrical complexes of protein subunits.
Dual polarisation interferometry
Dual polarisation interferometry provides complementary information about surface captured proteins. It assists in determining structure and conformation changes over time.
Projects
Prediction algorithm
The Folding@home project at Stanford University is a distributed computing research effort which uses approximately 5 petaFLOPS (≈10 x86 petaFLOPS) of available computing. It aims to find an algorithm which will consistently predict protein tertiary and quaternary structures given the protein's amino acid sequence and its cellular conditions.[9][10][11]
A list of software for protein tertiary structure prediction can be found at List of protein structure prediction software.
Protein aggregation diseases
Protein aggregation diseases such as Alzheimer's disease and Huntington's disease and prion diseases such as bovine spongiform encephalopathy can be better understood by constructing (and reconstructing) disease models. This is done by causing the disease in laboratory animals, for example, by administering a toxin, such as MPTP to cause Parkinson's disease, or through genetic manipulation.[12][13] Protein structure prediction is a new way to create disease models, which may avoid the use of animals.[14]
Protein Tertiary Structure Retrieval Project (CoMOGrad)
Matching patterns in tertiary structure of a given protein to huge number of known protein tertiary structures and retrieve most similar ones in ranked order is in the heart of many research areas like function prediction of novel proteins, study of evolution, disease diagnosis, drug discovery, antibody design etc. The CoMOGrad project at BUET is a research effort to device an extremely fast and much precise method for protein tertiary structure retrieval and develop online tool based on research outcome.[15][16]
See also
References
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "tertiary structure". doi:10.1351/goldbook.T06282
- Branden C. and Tooze J. "Introduction to Protein Structure" Garland Publishing, New York. 1990 and 1991.
- Kyte, J. "Structure in Protein Chemistry." Garland Publishing, New York. 1995. ISBN 0-8153-1701-8
- Senechal M. "I died for beauty: Dorothy Wrinch and the cultures of science." Oxford University Press, 2012. Chapter 14. ISBN 0-19-991083-9, 9780199910830. Accessed at Google Books 8 December 2013.
- Whisstock J (2006). "Molecular gymnastics: serpiginous structure, folding and scaffolding". Current Opinion in Structural Biology. 16 (6): 761–68. doi:10.1016/j.sbi.2006.10.005. PMID 17079131.
- Gettins PG (2002). "Serpin structure, mechanism, and function". Chem Rev. 102 (12): 4751–804. doi:10.1021/cr010170. PMID 12475206.
- Whisstock JC, Skinner R, Carrell RW, Lesk AM (2000). "Conformational changes in serpins: I. The native and cleaved conformations of alpha(1)-anti-trypsin". J Mol Biol. 296 (2): 685–99. doi:10.1006/jmbi.1999.3520. PMID 10669617.
- Seeliger, D; De Groot, B. L. (2010). "Conformational transitions upon ligand binding: Holo-structure prediction from apo conformations". PLOS Computational Biology. 6 (1): e1000634. Bibcode:2010PLSCB...6E0634S. doi:10.1371/journal.pcbi.1000634. PMC 2796265. PMID 20066034.
- "Folding@home." Stanford University. Accessed 18 December 2013.
- "Folding@home – FAQ" Stanford University. Accessed 18 December 2013.
- "Folding@home – Science." Stanford University.
- Schober A (October 2004). "Classic toxin-induced animal models of Parkinson's disease: 6-OHDA and MPTP". Cell Tissue Res. 318 (1): 215–24. doi:10.1007/s00441-004-0938-y. PMID 15503155. S2CID 1824912.
- "Tp53 Knockout Rat". Cancer. Retrieved 2010-12-18.
- "Feature – What is Folding and Why Does it Matter?". Archived from the original on December 12, 2013. Retrieved December 18, 2010.
- "Comograd :: Protein Tertiary Matching".
- Karim, Rezaul; Aziz, Mohd Momin Al; Shatabda, Swakkhar; Rahman, M. Sohel; Mia, Md Abul Kashem; Zaman, Farhana; Rakin, Salman (21 August 2015). "CoMOGrad and PHOG: From Computer Vision to Fast and Accurate Protein Tertiary Structure Retrieval". Scientific Reports. 5 (1): 13275. arXiv:1409.0814. Bibcode:2015NatSR...513275K. doi:10.1038/srep13275. PMC 4543952. PMID 26293226.
External links
- Protein Data Bank
- Display, analyse and superimpose protein 3D structures
- Alphabet of protein structures.
- Display, analyse and superimpose protein 3D structures
- WWW-based course teaching elementary protein bioinformatics
- Critical Assessment of Structure Prediction (CASP)
- Structural Classification of Proteins (SCOP)
- CATH Protein Structure Classification
- DALI/FSSP software and database of superposed protein structures
- TOPOFIT-DB Invariant Structural Cores between proteins
- PDBWiki — PDBWiki Home Page – a website for community annotation of PDB structures.