Tricarboxylate transport protein, mitochondrial
Tricarboxylate transport protein, mitochondrial, also known as tricarboxylate carrier protein and citrate transport protein (CTP), is a protein that in humans is encoded by the SLC25A1 gene.[3][4][5][6] SLC25A1 belongs to the mitochondrial carrier gene family SLC25.[7][8][9] High levels of the tricarboxylate transport protein are found in the liver, pancreas and kidney. Lower or no levels are present in the brain, heart, skeletal muscle, placenta and lung.[7][9]
| SLC25A1 | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Aliases | SLC25A1, CTP, D2L2AD, SEA, SLC20A3, solute carrier family 25 member 1, CMS23 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| External IDs | OMIM: 190315 HomoloGene: 136551 GeneCards: SLC25A1 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||

The tricarboxylate transport protein is located within the inner mitochondria membrane. It provides a link between the mitochondrial matrix and cytosol by transporting citrate through the impermeable inner mitochondrial membrane in exchange for malate from the cytosol.[7][8][9][10] The citrate transported out of the mitochondrial matrix by the tricarboxylate transport protein is catalyzed by citrate lyase to acetyl CoA, the starting material for fatty acid biosynthesis, and oxaloacetate.[8] As well, cytosolic NADPH + H+ necessary for fatty acid biosynthesis is generated in the reduction of oxaloacetate to malate and pyruvate by malate dehydrogenase and the malic enzyme.[9][11][12] For these reasons, the tricarboxylate transport protein is considered to play a key role in fatty acid synthesis.[8]
Structure
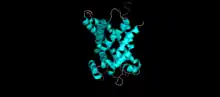
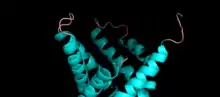

The structure of the tricarboxylate transport protein is consistent with the structures of other mitochondrial carriers.[7][8][10] In particular, the tricarboxylate transport protein has a tripartite structure consisting of three repeated domains that are approximately 100 amino acids in length.[7][10] Each repeat forms a transmembrane domain consisting of two hydrophobic α-helices.[7][8][13] The amino and carboxy termini are located on the cytosolic side of the inner mitochondrial membrane.[7][8] Each domain is linked by two hydrophilic loops located on the cytosolic side of the membrane.[7][8][13][14] The two α-helices of each repeated domain are connected by hydrophilic loops located on the matrix side of the membrane.[7][8][14] A salt bridge network is present on both the matrix side and cytoplasmic side of the tricarboxylate transport protein.[14]
Transport mechanism
The tricarboxylate transport protein exists in two states: a cytoplasmic state where it accepts malate from the cytoplasm and a matrix state where it accepts citrate from the mitochondrial matrix.[15] A single binding site is present near the center of the cavity of the tricarboxylate transport protein, which can be either exposed to the cytosol or the mitochondrial matrix depending on the state.[13][14][15] A substrate induced conformational change occurs when citrate enters from the matrix side and binds to the central cavity of the tricarboxylate transport protein.[7] This conformational change opens a gate on the cytosolic side and closes the gate on the matrix side.[7] Likewise, when malate enters from the cytosolic side, the matrix gate opens and the cytosolic gate closes.[7] Each side of the transporter is open and closed by the disruption and formation of the salt bridge networks, which allows access to the single binding site.[13][14][15][16][17]
Disease relevance
Mutations in this gene have been associated with the inborn error of metabolism combined D-2- and L-2-hydroxyglutaric aciduria,[18] which was the first reported case of a pathogenic mutation of the SLC25A1 gene.[14][19] Patients with D-2/L-2-hydroxyglutaric aciduria display neonatal onset metabolic encephalopathy, infantile epilepsy, global developmental delay, muscular hypotonia and early death.[14][19][20] It is believed low levels of citrate in the cytosol and high levels of citrate in the mitochondria caused by the impaired citrate transport plays a role in the disease.[14][20] In addition, increased expression of the tricarboxylate transport protein has been linked to cancer[9][21][22] and the production of inflammatory mediators.[23][24][25] Therefore, it has been suggested that inhibition of the tricarboxylate transport protein may have a therapeutic effect in chronic inflammation diseases and cancer.[24]
See also
- SLC25A1+protein,+human at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
References
- GRCh38: Ensembl release 89: ENSG00000100075 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Heisterkamp N, Mulder MP, Langeveld A, ten Hoeve J, Wang Z, Roe BA, Groffen J (September 1995). "Localization of the human mitochondrial citrate transporter protein gene to chromosome 22Q11 in the DiGeorge syndrome critical region". Genomics. 29 (2): 451–6. doi:10.1006/geno.1995.9982. PMID 8666394.
- Iacobazzi V, Lauria G, Palmieri F (September 1997). "Organization and sequence of the human gene for the mitochondrial citrate transport protein". DNA Sequence. 7 (3–4): 127–39. doi:10.3109/10425179709034029. PMID 9254007.
- Dolce V, Cappello AR, Capobianco L (September 1997). "Mitochondrial tricarboxylate and dicarboxylate-tricarboxylate carriers: from animals to plants". IUBMB Life. 66 (7): 462–71. doi:10.1002/iub.1290. PMID 25045044.
- "Entrez Gene: SLC25A1 solute carrier family 25 (mitochondrial carrier; citrate transporter), member 1".
- Palmieri F (April 2013). "The mitochondrial transporter family SLC25: identification, properties and physiopathology". Molecular Aspects of Medicine. 34 (2–3): 465–84. doi:10.1016/j.mam.2012.05.005. PMID 23266187.
- Palmieri F (February 2004). "The mitochondrial transporter family (SLC25): physiological and pathological implications". Pflügers Archiv. 447 (5): 689–709. doi:10.1007/s00424-003-1099-7. PMID 14598172. S2CID 25304722.
- Iacobazzi V, Infantino V, Palmieri F (January 2013). "Transcriptional Regulation of the Mitochondrial Citrate and Carnitine/Acylcarnitine Transporters: Two Genes Involved in Fatty Acid Biosynthesis and β-oxidation". Biology. 2 (1): 284–303. doi:10.3390/biology2010284. PMC 4009865. PMID 24832661.
- Berg JM, Tymoczko JL, Gatto GJ, Stryer L (2015). Biochemistry. New York: W.H. Freeman & Company. p. 551. ISBN 978-1-4641-2610-9.
- Voet D, Voet JG, Pratt CW (2016). Fundamentals of Biochemistry. U.S.A.: Wiley. pp. 687–688. ISBN 978-1-118-91840-1.
- Nelson DL, Cox MM (2017). Principles of Biochemistry. New York: W.H. Freeman & Company. pp. 818–819. ISBN 978-1-4641-2611-6.
- King MS, Kerr M, Crichton PG, Springett R, Kunji ER (January 2016). "Formation of a cytoplasmic salt bridge network in the matrix state is a fundamental step in the transport mechanism of the mitochondrial ADP/ATP carrier". Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1857 (1): 14–22. doi:10.1016/j.bbabio.2015.09.013. PMC 4674015. PMID 26453935.
- Majd H, King MS, Smith AC, Kunji ER (January 2018). "Pathogenic mutations of the human mitochondrial citrate carrier SLC25A1 lead to impaired citrate export required for lipid, dolichol, ubiquinone and sterol synthesis". Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1859 (1): 1–7. doi:10.1016/j.bbabio.2017.10.002. PMID 29031613.
- Robinson AJ, Kunji ER (February 2006). "Mitochondrial carriers in the cytoplasmic state have a common substrate binding site". Proceedings of the National Academy of Sciences of the United States of America. 103 (8): 2617–22. Bibcode:2006PNAS..103.2617R. doi:10.1073/pnas.0509994103. PMC 1413793. PMID 16469842.
- Robinson AJ, Overy C, Kunji ER (November 2008). "The mechanism of transport by mitochondrial carriers based on analysis of symmetry". Proceedings of the National Academy of Sciences of the United States of America. 105 (46): 17766–71. Bibcode:2008PNAS..10517766R. doi:10.1073/pnas.0809580105. PMC 2582046. PMID 19001266.
- Kunji ER, Robinson AJ (September 2006). "The conserved substrate binding site of mitochondrial carriers". Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1757 (9–10): 1237–48. doi:10.1016/j.bbabio.2006.03.021. PMID 16759636.
- Nota B, Struys EA, Pop A, Jansen EE, Fernandez Ojeda MR, Kanhai WA, Kranendijk M, van Dooren SJ, Bevova MR, Sistermans EA, Nieuwint AW, Barth M, Ben-Omran T, Hoffmann GF, de Lonlay P, McDonald MT, Meberg A, Muntau AC, Nuoffer JM, Parini R, Read MH, Renneberg A, Santer R, Strahleck T, van Schaftingen E, van der Knaap MS, Jakobs C, Salomons GS (April 2013). "Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined D-2- and L-2-hydroxyglutaric aciduria". American Journal of Human Genetics. 92 (4): 627–31. doi:10.1016/j.ajhg.2013.03.009. PMC 3617390. PMID 23561848.
- Hoffmann GF, Köckler S (2016). "Cerebral Organic Acid Disorders and Other Disorders of Lysine Catabolism". In Saudubray JM, Baumgartner M, Walter J (eds.). Inborn Metabolic Diseases. Germany: Springer. p. 344. ISBN 978-3-662-49771-5.
- Cohen I, Staretz-Chacham O, Wormser O, Perez Y, Saada A, Kadir R, Birk OS (February 2018). "A novel homozygous SLC25A1 mutation with impaired mitochondrial complex V: Possible phenotypic expansion". American Journal of Medical Genetics. Part A. 176 (2): 330–336. doi:10.1002/ajmg.a.38574. PMID 29226520. S2CID 6953669.
- Jiang L, Boufersaoui A, Yang C, Ko B, Rakheja D, Guevara G, Hu Z, DeBerardinis RJ (September 2017). "Quantitative metabolic flux analysis reveals an unconventional pathway of fatty acid synthesis in cancer cells deficient for the mitochondrial citrate transport protein". Metabolic Engineering. 43 (Pt B): 198–207. doi:10.1016/j.ymben.2016.11.004. PMC 5429990. PMID 27856334.
- Wan-angkan, P.; et al. (2018). "Combination of Mitochondrial and Plasma Membrane Citrate Transporter Inhibitors Inhibits De Novo Lipogenesis Pathway and Triggers Apoptosis in Hepatocellular Carcinoma Cells". BioMed Research International. 2018: 3683026. doi:10.1155/2018/3683026. PMC 5818947. PMID 29546056.
- Infantino V, Convertini P, Cucci L, Panaro MA, Di Noia MA, Calvello R, Palmieri F, Iacobazzi V (September 2011). "The mitochondrial citrate carrier: a new player in inflammation". The Biochemical Journal. 438 (3): 433–6. doi:10.1042/BJ20111275. PMID 21787310.
- Infantino V, Iacobazzi V, Menga A, Avantaggiati ML, Palmieri F (November 2014). "A key role of the mitochondrial citrate carrier (SLC25A1) in TNFα- and IFNγ-triggered inflammation". Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 1839 (11): 1217–1225. doi:10.1016/j.bbagrm.2014.07.013. PMC 4346166. PMID 25072865.
- Palmieri EM, Spera I, Menga A, Infantino V, Porcelli V, Iacobazzi V, Pierri CL, Hooper DC, Palmieri F, Castegna A (August 2015). "Acetylation of human mitochondrial citrate carrier modulates mitochondrial citrate/malate exchange activity to sustain NADPH production during macrophage activation". Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1847 (8): 729–38. doi:10.1016/j.bbabio.2015.04.009. PMID 25917893.
Further reading
- Ewing RM, Chu P, Elisma F, Li H, Taylor P, Climie S, McBroom-Cerajewski L, Robinson MD, O'Connor L, Li M, Taylor R, Dharsee M, Ho Y, Heilbut A, Moore L, Zhang S, Ornatsky O, Bukhman YV, Ethier M, Sheng Y, Vasilescu J, Abu-Farha M, Lambert JP, Duewel HS, Stewart II, Kuehl B, Hogue K, Colwill K, Gladwish K, Muskat B, Kinach R, Adams SL, Moran MF, Morin GB, Topaloglou T, Figeys D (2007). "Large-scale mapping of human protein-protein interactions by mass spectrometry". Molecular Systems Biology. 3 (1): 89. doi:10.1038/msb4100134. PMC 1847948. PMID 17353931.
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, Klitgord N, Simon C, Boxem M, Milstein S, Rosenberg J, Goldberg DS, Zhang LV, Wong SL, Franklin G, Li S, Albala JS, Lim J, Fraughton C, Llamosas E, Cevik S, Bex C, Lamesch P, Sikorski RS, Vandenhaute J, Zoghbi HY, Smolyar A, Bosak S, Sequerra R, Doucette-Stamm L, Cusick ME, Hill DE, Roth FP, Vidal M (October 2005). "Towards a proteome-scale map of the human protein-protein interaction network". Nature. 437 (7062): 1173–8. Bibcode:2005Natur.437.1173R. doi:10.1038/nature04209. PMID 16189514. S2CID 4427026.
- Gong W, Emanuel BS, Collins J, Kim DH, Wang Z, Chen F, Zhang G, Roe B, Budarf ML (June 1996). "A transcription map of the DiGeorge and velo-cardio-facial syndrome minimal critical region on 22q11". Human Molecular Genetics. 5 (6): 789–800. CiteSeerX 10.1.1.539.9441. doi:10.1093/hmg/5.6.789. PMID 8776594.
- Goldmuntz E, Wang Z, Roe BA, Budarf ML (April 1996). "Cloning, genomic organization, and chromosomal localization of human citrate transport protein to the DiGeorge/velocardiofacial syndrome minimal critical region". Genomics. 33 (2): 271–6. doi:10.1006/geno.1996.0191. PMID 8660975.
- Bonofiglio D, Santoro A, Martello E, Vizza D, Rovito D, Cappello AR, Barone I, Giordano C, Panza S, Catalano S, Iacobazzi V, Dolce V, Andò S (June 2013). "Mechanisms of divergent effects of activated peroxisome proliferator-activated receptor-γ on mitochondrial citrate carrier expression in 3T3-L1 fibroblasts and mature adipocytes". Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 1831 (6): 1027–36. doi:10.1016/j.bbalip.2013.01.014. hdl:11586/65706. PMID 23370576.
This article incorporates text from the United States National Library of Medicine, which is in the public domain.