Discovery and development of gliflozins
Gliflozins are a class of drugs in the treatment of type 2 diabetes (T2D). They act by inhibiting sodium/glucose cotransporter 2 (SGLT-2), and are therefore also called SGLT-2 inhibitors. The efficacy of the drug is dependent on renal excretion and prevents glucose from going into blood circulation by promoting glucosuria. The mechanism of action is insulin independent.
Three drugs have been accepted by the Food and Drug Administration (FDA) in the United States; dapagliflozin, canagliflozin and empagliflozin. Canagliflozin was the first SGLT-2 inhibitor that was approved by the FDA, being accepted in March 2013. Dapagliflozin and empagliflozin were accepted in 2014.
Introduction
Role of kidneys in glucose homeostasis
There are at least four members of SLC-5 gene family, which are secondary active glucose transporters. The sodium glucose transporters proteins SGLT-1 and SGLT-2 are the two premier members of the family. These two members are found in the kidneys, among other transporters, and are the main co-transporters there related to the blood sugar. They play a role in renal glucose reabsorption and in intestinal glucose absorption.[1][2]
Blood glucose is freely filtered by the glomeruli and SGLT-1 and SGLT-2 reabsorb glucose in the kidneys and put it back into the circulation cells. SGLT-2 is responsible for 90% of the reabsorption and SGLT-1 for the other 10%.[1][3]
SGLT-2 protein
Sodium/glucose co-transporter (SGLT) proteins are bound to the cell membrane and have the role of transporting glucose through the membrane into the cells, against the concentration gradient of glucose. This is done by using the sodium gradient, produced by sodium/potassium ATPase pumps, so at the same time glucose is transported into the cells, the sodium is too. Since it is against the gradient, it requires energy to work. SGLT proteins cause the glucose reabsorption from the glomerular filtrate, independent of insulin.[1][3][4]
SGLT-2 is a member of the glucose transporter family and is a low-affinity, high-capacity glucose transporter. SGLT-2 is mainly expressed in the S-1 and S-2 segments of the proximal renal tubules where the majority of filtered glucose is absorbed. SGLT-2 has a role in regulation of glucose and is responsible for most glucose reabsorption in the kidneys.[1][5]
In diabetes, extracellular glucose concentration increases and this high glucose level leads to upregulation of SGLT-2, leading in turn to more absorption of glucose in the kidneys. These effects cause maintenance of hyperglycemia.[6] Because sodium is absorbed at the same time as glucose via SGLT-2, the upregulation of SGLT-2 probably leads to development or maintenance of hypertension. In study where rats were given either ramipril or losartan, levels of SGLT-2 protein and mRNA were significantly reduced. In patients with diabetes, hypertension is a common problem so this may have relevance in this disease.[1]
Drugs that inhibit sodium/glucose cotransporter 2 inhibit renal glucose reabsorption which leads to enhanced urinary glucose excretion and lower glucose in blood. They work independently of insulin and can reduce glucose levels without causing hypoglycemia or weight gain.[1][7]
Discovery
Medieval physicians routinely tasted urine and wrote discourses on their observations. Which physician originally thought that diabetes mellitus was a renal disorder because of glucose discharged in urine is apparently now lost to history. The discovery of insulin eventually led to a diabetes management focus on the pancreas. Traditional foci of therapeutic strategies for diabetes have been on enhancing endogenous insulin secretion and on improving insulin sensitivity.
In the previous decade the role of the kidney in the development and maintenance of high glucose levels has been examined. The role of the kidney led to the development of drugs that inhibit the sodium/glucose transporter 2 protein. Every day approximately 180 grams of glucose are filtered through the glomeruli and lost into the primary urine in healthy adults, but more than 90% of the glucose that is initially filtered is reabsorbed by a high capacity system controlled by SGLT-2 in the early convoluted segment of the proximal tubules. Almost all remaining filtered glucose is reabsorbed by sodium/glucose transporter 1 so under normal circumstances almost all filtered glucose will be reabsorbed and less than 100 mg of glucose finds its way into the urine of non-diabetic individuals.[8][9]
Phlorizin
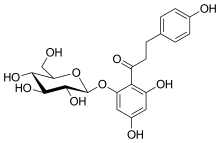
Phlorizin is a compound that has been known for over a century. It is a naturally occurring botanical glucoside that produces renal glucosuria and blocks intestinal glucose absorption through inhibition of the sodium/glucose symporters located in the proximal renal tubule and mucosa of the small intestine. Phlorizin was first isolated in 1835 and was subsequently found to be a potent but rather non-selective inhibitor of both SGLT-1 and SGLT-2 proteins.[10][11][12]
Phlorizin seemed to have very interesting properties and the results in animal studies were encouraging, it improved insulin sensitivity and in diabetic rat models it seemed to increase glucose levels in urine and also normal glucose concentration in plasma occurred without hypoglycemia. Unfortunately, in spite of these properties, phlorizin was not suitable enough for clinical development for several reasons. Phlorizin has very poor oral bioavailability as it is broken down in the gastrointestinal tract, so it has to be given parenterally. Phloretin, the active metabolite of phlorizin, is a potent inhibitor of facilitative glucose transporters and phlorizin seems to lead to serious adverse events in the gastrointestinal tract like diarrhea and dehydration. Because of these reasons, phlorizin was never pursued in humans.[10][12][13]
Although phlorizin was not suitable for further clinical trials, it served an important role in the development of SGLT-2 inhibitors. It served a basis for the recognition of SGLT inhibitors with improved safety and tolerability profiles. For an example, the SGLT inhibitors are not associated with gastrointestinal adverse events and the bioavailability is much greater.[4][12][13]
Inhibition of SGLT-2 results as better control of glucose level, lower insulin, lower blood pressure and uric acid levels and augments calorie wasting. Some data supports the hypothesis that SGLT-2 inhibition may have direct renoprotective effects. This includes actions to attenuate tubular hypertrophy and hyperfiltration associated with diabetes and to reduce the tubular toxicity of glucose. Inhibition of SGLT-2 following treatment with dapagliflozin reduces the capacity for tubular glucose reabsorption by approximately 30–50%.[8]
Drug development
Phlorizin consists of glucose moiety and two aromatic rings (aglycone moiety) joined by an alkyl spacer. Initially, phlorizin was isolated for treatment of fever and infectious diseases, particularly malaria. According to Michael Nauck and his partners, studies were made in the 1950s on phlorizin that showed that it could block sugar transport in the kidney, small intestine, and a few other tissues. In the early 1990s, sodium/glucose cotransporter 2 was fully characterized, so the mechanism of phlorizin became of real interest. In later studies it was said that sugar-blocking effects of phlorizin was due to inhibition of the sodium/glucose cotransporter proteins.[1][4][9][11]
Most of the reported SGLT-2 inhibitors are glucoside analogs that can be tracked to the o-aryl glucoside found in the nature. The problem with using o-glucosides as SGLT-2 inhibitors is instability that can be tracked to degradation by β-glucosidase in the small intestine. Because of that, o-glucosides given orally have to be prodrug esters. These prodrugs go through changes in the body leading to carbon–carbon bond between the glucose and the aglycone moiety so c-glucoside are formed from the o-glucosides. C-glucosides have a different pharmacokinetic profile than o-glucosides (e.g. half-life and duration of action) and are not degraded by the β-glucosidase. The first discovered c-glucoside was the drug dapagliflozin.[1][14][15] Dapagliflozin was the first highly selective SGLT-2-inhibitor approved by the European Medicines Agency.[16] All SGLT-2 inhibitors in clinical development are prodrugs that have to be converted to its active ‘A’ form for activity.[9]
T-1095
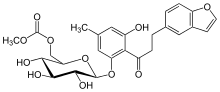
Because Phlorizin is a nonselective inhibitor with poor oral bioavailability, a phlorizin derivative was synthesised and called T-1095. T-1095 is a methyl carbonate prodrug that is absorbed into the circulation when given orally, and is rapidly converted in the liver to the active metabolite T-1095A.[1][9] By inhibiting SGLT-1 and SGLT-2, urinary glucose excretion increased in diabetic animals. T-1095 did not proceed in clinical development, probably because of the inhibition of SGLT-1[1] but non-selective SGLT inhibitors may also block glucose transporter 1 (GLUT-1). Because 90% of filtered glucose is reabsorbed through SGLT-2, research has focused specifically on SGLT-2. Inhibition of SGLT-1 may also lead to the genetic disease glucose-galactose malabsorption, which is characterized by severe diarrhea.[9][17]
ISIS 388626
According to preliminary findings of a novel method of SGLT-2 inhibition, the antisense oligonucleotide ISIS 388626 improved plasma glucose in rodents and dogs by reducing mRNA expression in the proximal renal tubules by up to 80% when given once a week. It did not affect SGLT-1. A study results on long-term use of ISIS 388626 in non-human primates observed more than 1000 fold increase in glucosuria without any associated hypoglycemia. This increase in glucosuria can be attributed to a dose-dependent reduction in the expression of SGLT-2, where the highest dose led to more than 75% reduction.[9] In 2011, Ionis Pharmaceuticals initiated a clinical phase 1 study with ISIS-SGLT-2RX, a 12-nucleotide antisense oligonucleotide.[18] Results from this study were published in 2017 and the treatment was "associated with unexpected renal effects". The authors concluded that "Before the concept of antisense-mediated blocking of SGLT2 with ISIS 388626 can be explored further, more preclinical data are needed to justify further investigations."[19]
Activity of SGLT-2 inhibitors in glycemic control
Michael Nauck recounts that meta-analyses of studies about the activity of SGLT-2 inhibitors in glycemic control in type 2 diabetes mellitus patients shows improvement in the control of glucose, when compared with placebos, metformin, sulfonylurea, thiazolidinediones, insulin and more. The HbA1c was examined after SGLT-2 inhibitors were given alone (as monotherapy) and as an add-on therapy to the other diabetes medicines. The SGLT-2 inhibitors that were used were dapagliflozin and canagliflozin and others in the same drug class. The meta-analysis was taken together from studies ranging from period of few weeks up to more than 100 weeks.[4]
The results, summed up, were that 10 mg of dapagliflozin showed more effect than placebo in the control of glucose, when given for 24 weeks. However, no inferior efficacy of 10 mg dapagliflozin was shown when used as an add-on therapy to metformin, compared with glipizide after use for 52 weeks. 10 mg of dapagliflozin showed neither inferior efficacy compared with metformin when both of the medicines were given as monotherapy for 24 weeks.[4]
The results from meta-analysis when canagliflozin was examined, showed that compared to a placebo, canagliflozin affects HbA1c. Meta-analysis studies also showed that 10 mg and 25 mg of empagliflozin, improved HbA1c compared with a placebo.[4]
Structure-activity relationship (SAR)
The aglycones of both phlorizin and dapagliflozin have weak inhibition effects on SGLT-1 and SGLT-2. Two synergistic forces are involved in binding of inhibitors to SGLTs. Different sugars on the aglycone will affect and change the orientation of it in the access vestibule because one of the forces involved in the binding is the binding of sugar to the glucose site. The other force is the binding of the aglycone, which affects the binding affinity of the entire inhibitor.[14]
The discovery of T-1095 led to an investigation of how to enhance potency, selectivity and oral bioavailability by adding various substituents to the glycoside core. As an example we can take the change of o-glycosides to c-glycosides by creating a carbon–carbon bond between the glucose and the aglycone moiety. C-glucosides are more stable than o-glucosides which leads to modified half-life and duration of action. These modifications have also led to more specificity to SGLT-2.[9] C-glucosides that have heterocyclic ring at the distal ring or proximal ring are better when it comes to anti-diabetic effect and physicochemical features all together.[3][20] C-glucoside bearing thiazole at the distal ring on canagliflozin has shown good physicochemical properties that can lead to a clinical development, but still has the same anti-diabetic activity as dapagliflozin, as shown in tables 1 and 2.
Song and his partners did preparate thiazole compound by starting with carboxyl acid. Working with that, it took them three steps to get a compound like dapagliflozin with a thiazole ring. Inhibitory effects on SGLT-2 of the compounds were tested by Song and his partners. In tables 1, 2, and 3, the IC50 value changes depending on what compound is in the ring position, in the C-4 region of the proximal phenyl ring, and how the thiazole ring relates.[3]
Many compounds gave different IC50 value in the ring position in an in vitro activity. For an example there was a big difference if there was an n-pentyl group (IC50 = 13,3 nM), n-butyl (IC50 = 119 nM), phenyl with 2-furyl (IC50 = 0,720) or 3-thiophenyl (IC50 = 0,772). As seen in table 1, the in vitro activity increases depending on what compound is bonded to the distal ring (given that in the C-4 region of the proximal phenyl ring is a Cl atom).[3]
Table 1: Differences in in vitro activity depending on which compound is bonded to the distal ring.
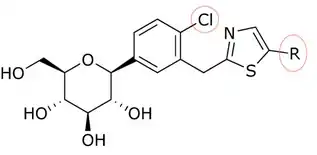
| R | IC50 (nM)[3] | Activity[3] |
|---|---|---|
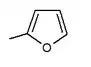 | 0.720 | in vitro activity improved* |
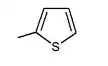 | 1.14 | in vitro activity improved* |
| 13.3 | As the number of carbons increases, the IC-50 value fluctuates | |
| 19.6 | in vitro activity decreased* | |
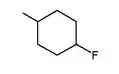 | 21.2 | in vitro activity decreased* |
*comparator to ethyl group (IC50 = 16,7)
In table 2, the in vitro activity changes depending on the compound in the C-4 region of the proximal phenyl ring (X). Small methyl groups or other halogen atoms in the C-4 position gave IC50 ranging from 0.72–36.7 (given that the phenyl with 2-furyl is in the ring position).[3]
Table 2: Differences in in vitro activity depending on what compound is in the C-4 region of the proximal phenyl ring.
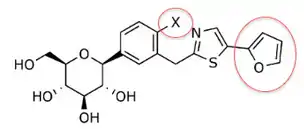
| X[3] | IC50 (nM)[3] |
|---|---|
| Cl | 0.720 |
| Me | 1.43 |
| F | 6.11 |
| H | 22.6 |
| CN | 36.7 |
Table 3: Difference in the IC50 value depending on how the thiazole ring relates (nothing else is changed in the structure (X = Cl, R = phenyl with 2-furyl).
| Compound | IC50 (nM)[3] |
|---|---|
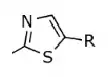 | 0.720 |
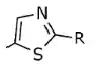 | 1.11 |
See also
- Sodium-glucose transport proteins
- SLC5A2
- SGLT1
- SGLT2
- Dapagliflozin
- Empagliflozin
- Canagliflozin
- Ipragliflozin
References
- Nair, S.; Wilding, J.P.H. (2010). "Sodium Glucose Cotransporter 2 Inhibitors as a New Treatment for Diabetes Mellitus". Journal of Clinical Endocrinology & Metabolism. 95 (1): 34–42. doi:10.1210/jc.2009-0473. PMID 19892839.
- Wright, E.M.; Hirayama, B.A.; Loo, D.F. (2007). "Active sugar transport in health and disease". Journal of Internal Medicine. 261 (1): 32–43. doi:10.1111/j.1365-2796.2006.01746.x. PMID 17222166.
- Song, Kwang-Seop; Lee, Suk Ho; Kim, Min Ju; Seo, Hee Jeong; Lee, Junwon; Lee, Sung-Han; Jung, Myung Eun; Son, Eun-Jung; Lee, MinWoo; Kim, Jeongmin; Lee, Jinhwa (10 February 2011). "Synthesis and SAR of Thiazolylmethylphenyl Glucoside as Novel-Aryl Glucoside SGLT2 Inhibitors". ACS Medicinal Chemistry Letters. 2 (2): 182–187. doi:10.1021/Ml100256c. PMC 4018110. PMID 24900297.
- Nauck, Michael (2014). "Update on developments with SGLT2 inhibitors in the management of type 2 diabetes". Drug Design, Development and Therapy. 8: 1335–1380. doi:10.2147/DDDT.S50773. PMC 4166348. PMID 25246775.
- Kasichayanula, Sreeneeranj; Liu, Xiaoni; Pe Benito, Melanie; Yao, Ming; Pfister, Marc; LaCreta, Frank P.; Humphreys, William Griffith; Boulton, David W. (2013). "The influence of kidney function on dapagliflozin exposure, metabolism and pharmacodynamics in healthy subjects and in patients with type 2 diabetes mellitus". British Journal of Clinical Pharmacology. 76 (3): 432–444. doi:10.1111/bcp.12056. PMC 3769670. PMID 23210765.
- Mediavilla Bravo, J.J. (July 2014). "Aportaciones de los SGLT-2 y nuevos fármacos en investigación". SEMERGEN - Medicina de Familia. 40: 34–40. doi:10.1016/S1138-3593(14)74388-6. PMID 25311718.
- Whaley, Jean; Tirmenstein; Reilly; Poucher; Saye; Parikh; List (2012). "Targeting the kidney and glucose excretion with dapagliflozin: preclinical and clinical evidence for SGLT2 inhibition as a new option for treatment of type 2 diabetes mellitus". Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy. 5: 135–48. doi:10.2147/DMSO.S22503. PMC 3422910. PMID 22923998.
- Thomas, M. C. (24 July 2014). "Renal effects of dapagliflozin in patients with type 2 diabetes". Therapeutic Advances in Endocrinology and Metabolism. 5 (3): 53–61. doi:10.1177/2042018814544153. PMC 4132377. PMID 25126408.
- Idris, Iskandar; Donnelly, Richard (2009). "Sodium-glucose co-transporter-2 inhibitors: an emerging new class of oral antidiabetic drug". Diabetes, Obesity and Metabolism. 11 (2): 79–88. doi:10.1111/j.1463-1326.2008.00982.x. PMID 19125776.
- Ehrenkranz, Joel R. L.; Lewis, Norman G.; Ronald Kahn, C.; Roth, Jesse (2005). "Phlorizin: a review". Diabetes/Metabolism Research and Reviews. 21 (1): 31–38. doi:10.1002/dmrr.532. PMID 15624123. S2CID 37909306.
- Chao, Edward C.; Henry, Robert R. (28 May 2010). "SGLT2 inhibition — a novel strategy for diabetes treatment". Nature Reviews Drug Discovery. 9 (7): 551–559. doi:10.1038/nrd3180. PMID 20508640. S2CID 21788174.
- McGill, Janet B. (12 April 2014). "The SGLT2 Inhibitor Empagliflozin for the Treatment of Type 2 Diabetes Mellitus: a Bench to Bedside Review". Diabetes Therapy. 5 (1): 43–63. doi:10.1007/s13300-014-0063-1. PMC 4065283. PMID 24729157.
- Bhartia, Mithun; Tahrani, Abd A.; Barnett, Anthony H. (2011). "SGLT-2 Inhibitors in Development for Type 2 Diabetes Treatment". The Review of Diabetic Studies. 8 (3): 348–354. doi:10.1900/RDS.2011.8.348. PMC 3280669. PMID 22262072.
- Hummel, C. S.; Lu, C.; Liu, J.; Ghezzi, C.; Hirayama, B. A.; Loo, D. D. F.; Kepe, V.; Barrio, J. R.; Wright, E. M. (21 September 2011). "Structural selectivity of human SGLT inhibitors". AJP: Cell Physiology. 302 (2): C373–C382. doi:10.1152/ajpcell.00328.2011. PMC 3328840. PMID 21940664.
- Li, An-Rong; Zhang, Jian; Greenberg, Joanne; Lee, TaeWeon; Liu, Jiwen (2011). "Discovery of non-glucoside SGLT2 inhibitors". Bioorganic & Medicinal Chemistry Letters. 21 (8): 2472–2475. doi:10.1016/j.bmcl.2011.02.056. PMID 21398124.
- Cuypers, J; Mathieu, C; Benhalima, K (2013). "SGLT2-inhibitors: a novel class for the treatment of type 2 diabetes introduction of SGLT-2-inhibitors in clinical practice". Acta Clinica Belgica. 68 (4): 287–93. doi:10.2143/acb.3349. PMID 24455799. S2CID 22590286.
- Lv, Binhua; Xu, Baihua; Feng, Yan; Peng, Kun; Xu, Ge; Du, Jiyan; Zhang, Lili; Zhang, Wenbin; Zhang, Ting; Zhu, Liangcheng; Ding, Haifeng; Sheng, Zelin; Welihinda, Ajith; Seed, Brian; Chen, Yuanwei (December 2009). "Exploration of O-spiroketal C-arylglucosides as novel and selective renal sodium-dependent glucose co-transporter 2 (SGLT2) inhibitors". Bioorganic & Medicinal Chemistry Letters. 19 (24): 6877–6881. doi:10.1016/j.bmcl.2009.10.088. PMID 19896374.
- "Isis Initiates Phase 1 Clinical Trial of SGLT2 Antisense Drug".
- van Meer L, van Dongen M, Moerland M, de Kam M, Cohen A, Burggraaf J (February 2017). "Novel SGLT2 inhibitor: first-in-man studies of antisense compound is associated with unexpected renal effects". Pharmacol Res Perspect. 5 (1): e00292. doi:10.1002/prp2.292. PMC 5461644. PMID 28596840.
- Park, Eun-Jung; Kong, Younggyu; Lee, Jun Sung; Lee, Sung-Han; Lee, Jinhwa (January 2011). "Exploration of SAR regarding glucose moiety in novel C-aryl glucoside inhibitors of SGLT2". Bioorganic & Medicinal Chemistry Letters. 21 (2): 742–746. doi:10.1016/J.Bmcl.2010.11.115. PMID 21193308.