Drug discovery
In the fields of medicine, biotechnology and pharmacology, drug discovery is the process by which new candidate medications are discovered.[1]
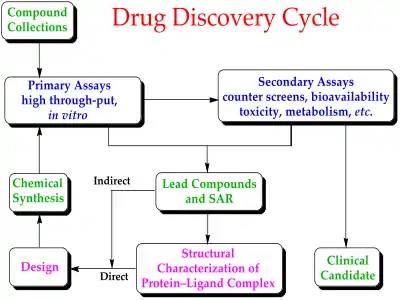
Historically, drugs were discovered by identifying the active ingredient from traditional remedies or by serendipitous discovery, as with penicillin. More recently, chemical libraries of synthetic small molecules, natural products or extracts were screened in intact cells or whole organisms to identify substances that had a desirable therapeutic effect in a process known as classical pharmacology. After sequencing of the human genome allowed rapid cloning and synthesis of large quantities of purified proteins, it has become common practice to use high throughput screening of large compounds libraries against isolated biological targets which are hypothesized to be disease-modifying in a process known as reverse pharmacology. Hits from these screens are then tested in cells and then in animals for efficacy.[2]
Modern drug discovery involves the identification of screening hits,[3] medicinal chemistry[4] and optimization of those hits to increase the affinity, selectivity (to reduce the potential of side effects), efficacy/potency, metabolic stability (to increase the half-life), and oral bioavailability. Once a compound that fulfills all of these requirements has been identified, the process of drug development can continue. If successful, clinical trials are developed.[5]
Modern drug discovery is thus usually a capital-intensive process that involves large investments by pharmaceutical industry corporations as well as national governments (who provide grants and loan guarantees). Despite advances in technology and understanding of biological systems, drug discovery is still a lengthy, "expensive, difficult, and inefficient process" with low rate of new therapeutic discovery.[6] In 2010, the research and development cost of each new molecular entity was about US$1.8 billion.[7] In the 21st century, basic discovery research is funded primarily by governments and by philanthropic organizations, while late-stage development is funded primarily by pharmaceutical companies or venture capitalists.[8] To be allowed to come to market, drugs must undergo several successful phases of clinical trials, and pass through a new drug approval process, called the New Drug Application in the United States.
Discovering drugs that may be a commercial success, or a public health success, involves a complex interaction between investors, industry, academia, patent laws, regulatory exclusivity, marketing and the need to balance secrecy with communication.[9] Meanwhile, for disorders whose rarity means that no large commercial success or public health effect can be expected, the orphan drug funding process ensures that people who experience those disorders can have some hope of pharmacotherapeutic advances.
History
The idea that the effect of a drug in the human body is mediated by specific interactions of the drug molecule with biological macromolecules, (proteins or nucleic acids in most cases) led scientists to the conclusion that individual chemicals are required for the biological activity of the drug. This made for the beginning of the modern era in pharmacology, as pure chemicals, instead of crude extracts of medicinal plants, became the standard drugs. Examples of drug compounds isolated from crude preparations are morphine, the active agent in opium, and digoxin, a heart stimulant originating from Digitalis lanata. Organic chemistry also led to the synthesis of many of the natural products isolated from biological sources.
Historically, substances, whether crude extracts or purified chemicals, were screened for biological activity without knowledge of the biological target. Only after an active substance was identified was an effort made to identify the target. This approach is known as classical pharmacology, forward pharmacology,[10] or phenotypic drug discovery.[11]
Later, small molecules were synthesized to specifically target a known physiological/pathological pathway, avoiding the mass screening of banks of stored compounds. This led to great success, such as the work of Gertrude Elion and George H. Hitchings on purine metabolism,[12][13] the work of James Black[14] on beta blockers and cimetidine, and the discovery of statins by Akira Endo.[15] Another champion of the approach of developing chemical analogues of known active substances was Sir David Jack at Allen and Hanbury's, later Glaxo, who pioneered the first inhaled selective beta2-adrenergic agonist for asthma, the first inhaled steroid for asthma, ranitidine as a successor to cimetidine, and supported the development of the triptans.[16]
Gertrude Elion, working mostly with a group of fewer than 50 people on purine analogues, contributed to the discovery of the first anti-viral; the first immunosuppressant (azathioprine) that allowed human organ transplantation; the first drug to induce remission of childhood leukemia; pivotal anti-cancer treatments; an anti-malarial; an anti-bacterial; and a treatment for gout.
Cloning of human proteins made possible the screening of large libraries of compounds against specific targets thought to be linked to specific diseases. This approach is known as reverse pharmacology and is the most frequently used approach today.[17]
Targets
A "target" is produced within the pharmaceutical industry.[8] Generally, the "target" is the naturally existing cellular or molecular structure involved in the pathology of interest where the drug-in-development is meant to act.[8] However, the distinction between a "new" and "established" target can be made without a full understanding of just what a "target" is. This distinction is typically made by pharmaceutical companies engaged in the discovery and development of therapeutics.[8] In an estimate from 2011, 435 human genome products were identified as therapeutic drug targets of FDA-approved drugs.[18]
"Established targets" are those for which there is a good scientific understanding, supported by a lengthy publication history, of both how the target functions in normal physiology and how it is involved in human pathology.[2] This does not imply that the mechanism of action of drugs that are thought to act through a particular established target is fully understood.[2] Rather, "established" relates directly to the amount of background information available on a target, in particular functional information. In general, "new targets" are all those targets that are not "established targets" but which have been or are the subject of drug discovery efforts. The majority of targets selected for drug discovery efforts are proteins, such as G-protein-coupled receptors (GPCRs) and protein kinases.[19]
Screening and design
The process of finding a new drug against a chosen target for a particular disease usually involves high-throughput screening (HTS), wherein large libraries of chemicals are tested for their ability to modify the target. For example, if the target is a novel GPCR, compounds will be screened for their ability to inhibit or stimulate that receptor (see antagonist and agonist): if the target is a protein kinase, the chemicals will be tested for their ability to inhibit that kinase.[20]
Another function of HTS is to show how selective the compounds are for the chosen target, as one wants to find a molecule which will interfere with only the chosen target, but not other, related targets.[20] To this end, other screening runs will be made to see whether the "hits" against the chosen target will interfere with other related targets – this is the process of cross-screening.[20] Cross-screening is useful because the more unrelated targets a compound hits, the more likely that off-target toxicity will occur with that compound once it reaches the clinic.[20]
It is unlikely that a perfect drug candidate will emerge from these early screening runs. One of the first steps is to screen for compounds that are unlikely to be developed into drugs; for example compounds that are hits in almost every assay, classified by medicinal chemists as "pan-assay interference compounds", are removed at this stage, if they were not already removed from the chemical library.[21][22][23] It is often observed that several compounds are found to have some degree of activity, and if these compounds share common chemical features, one or more pharmacophores can then be developed. At this point, medicinal chemists will attempt to use structure–activity relationships (SAR) to improve certain features of the lead compound:
- increase activity against the chosen target
- reduce activity against unrelated targets
- improve the druglikeness or ADME properties of the molecule.
This process will require several iterative screening runs, during which, it is hoped, the properties of the new molecular entities will improve, and allow the favoured compounds to go forward to in vitro and in vivo testing for activity in the disease model of choice.
Amongst the physicochemical properties associated with drug absorption include ionization (pKa), and solubility; permeability can be determined by PAMPA and Caco-2. PAMPA is attractive as an early screen due to the low consumption of drug and the low cost compared to tests such as Caco-2, gastrointestinal tract (GIT) and Blood–brain barrier (BBB) with which there is a high correlation.
A range of parameters can be used to assess the quality of a compound, or a series of compounds, as proposed in the Lipinski's Rule of Five. Such parameters include calculated properties such as cLogP to estimate lipophilicity, molecular weight, polar surface area and measured properties, such as potency, in-vitro measurement of enzymatic clearance etc. Some descriptors such as ligand efficiency[24] (LE) and lipophilic efficiency[25][26] (LiPE) combine such parameters to assess druglikeness.
While HTS is a commonly used method for novel drug discovery, it is not the only method. It is often possible to start from a molecule which already has some of the desired properties. Such a molecule might be extracted from a natural product or even be a drug on the market which could be improved upon (so-called "me too" drugs). Other methods, such as virtual high throughput screening, where screening is done using computer-generated models and attempting to "dock" virtual libraries to a target, are also often used.[20]
Another method for drug discovery is de novo drug design, in which a prediction is made of the sorts of chemicals that might (e.g.) fit into an active site of the target enzyme. For example, virtual screening and computer-aided drug design are often used to identify new chemical moieties that may interact with a target protein.[27][28] Molecular modelling[29] and molecular dynamics simulations can be used as a guide to improve the potency and properties of new drug leads.[30][31][32]
There is also a paradigm shift in the drug discovery community to shift away from HTS, which is expensive and may only cover limited chemical space, to the screening of smaller libraries (maximum a few thousand compounds). These include fragment-based lead discovery (FBDD)[33][34][35][36] and protein-directed dynamic combinatorial chemistry.[37][38][39][40][41] The ligands in these approaches are usually much smaller, and they bind to the target protein with weaker binding affinity than hits that are identified from HTS. Further modifications through organic synthesis into lead compounds are often required. Such modifications are often guided by protein X-ray crystallography of the protein-fragment complex.[42][43][44] The advantages of these approaches are that they allow more efficient screening and the compound library, although small, typically covers a large chemical space when compared to HTS.
Phenotypic screens have also provided new chemical starting points in drug discovery.[45][46] A variety of models have been used including yeast, zebrafish, worms, immortalized cell lines, primary cell lines, patient-derived cell lines and whole animal models. These screens are designed to find compounds which reverse a disease phenotype such as death, protein aggregation, mutant protein expression, or cell proliferation as examples in a more holistic cell model or organism. Smaller screening sets are often used for these screens, especially when the models are expensive or time-consuming to run.[47] In many cases, the exact mechanism of action of hits from these screens is unknown and may require extensive target deconvolution experiments to ascertain. The growth of the field of chemoproteomics has provided numerous strategies to identify drug targets in these cases.[48]
Once a lead compound series has been established with sufficient target potency and selectivity and favourable drug-like properties, one or two compounds will then be proposed for drug development. The best of these is generally called the lead compound, while the other will be designated as the "backup".[49] These decisions are generally supported by computational modelling innovations.[50][51][52]
Nature as source
Traditionally, many drugs and other chemicals with biological activity have been discovered by studying chemicals that organisms create to affect the activity of other organisms for survival.[53]
Despite the rise of combinatorial chemistry as an integral part of lead discovery process, natural products still play a major role as starting material for drug discovery.[54] A 2007 report[55] found that of the 974 small molecule new chemical entities developed between 1981 and 2006, 63% were natural derived or semisynthetic derivatives of natural products. For certain therapy areas, such as antimicrobials, antineoplastics, antihypertensive and anti-inflammatory drugs, the numbers were higher. In many cases, these products have been used traditionally for many years.
Natural products may be useful as a source of novel chemical structures for modern techniques of development of antibacterial therapies.[56]
Plant-derived
Many secondary metabolites produced by plants have potential therapeutic medicinal properties. These secondary metabolites contain, bind to, and modify the function of proteins (receptors, enzymes, etc.). Consequently, plant derived natural products have often been used as the starting point for drug discovery.[57][58][59][60][3]
History
Until the Renaissance, the vast majority of drugs in Western medicine were plant-derived extracts.[61] This has resulted in a pool of information about the potential of plant species as important sources of starting materials for drug discovery.[62] Botanical knowledge about different metabolites and hormones that are produced in different anatomical parts of the plant (e.g. roots, leaves, and flowers) are crucial for correctly identifying bioactive and pharmacological plant properties.[62][63] Identifying new drugs and getting them approved for market has proved to be a stringent process due to regulations set by national drug regulatory agencies.[64]
Jasmonates
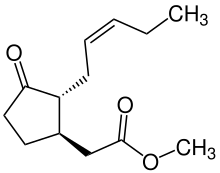
Jasmonates are important in responses to injury and intracellular signals. They induce apoptosis[65][66] and protein cascade via proteinase inhibitor,[65] have defense functions,[67] and regulate plant responses to different biotic and abiotic stresses.[67][68] Jasmonates also have the ability to directly act on mitochondrial membranes by inducing membrane depolarization via release of metabolites.[69]
Jasmonate derivatives (JAD) are also important in wound response and tissue regeneration in plant cells. They have also been identified to have anti-aging effects on human epidermal layer.[70] It is suspected that they interact with proteoglycans (PG) and glycosaminoglycan (GAG) polysaccharides, which are essential extracellular matrix (ECM) components to help remodel the ECM.[71] The discovery of JADs on skin repair has introduced newfound interest in the effects of these plant hormones in therapeutic medicinal application.[70]
Salicylates
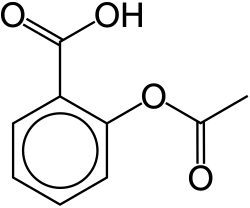
Salicylic acid (SA), a phytohormone, was initially derived from willow bark and has since been identified in many species. It is an important player in plant immunity, although its role is still not fully understood by scientists.[72] They are involved in disease and immunity responses in plant and animal tissues. They have salicylic acid binding proteins (SABPs) that have shown to affect multiple animal tissues.[72] The first discovered medicinal properties of the isolated compound was involved in pain and fever management. They also play an active role in the suppression of cell proliferation.[65] They have the ability to induce death in lymphoblastic leukemia and other human cancer cells.[65] One of the most common drugs derived from salicylates is aspirin, also known as acetylsalicylic acid, with anti-inflammatory and anti-pyretic properties.[72][73]
Microbial metabolites
Microbes compete for living space and nutrients. To survive in these conditions, many microbes have developed abilities to prevent competing species from proliferating. Microbes are the main source of antimicrobial drugs. Streptomyces isolates have been such a valuable source of antibiotics, that they have been called medicinal molds. The classic example of an antibiotic discovered as a defense mechanism against another microbe is penicillin in bacterial cultures contaminated by Penicillium fungi in 1928.
Marine invertebrates
Marine environments are potential sources for new bioactive agents.[74] Arabinose nucleosides discovered from marine invertebrates in 1950s, demonstrated for the first time that sugar moieties other than ribose and deoxyribose can yield bioactive nucleoside structures. It took until 2004 when the first marine-derived drug was approved. For example, the cone snail toxin ziconotide, also known as Prialt treats severe neuropathic pain. Several other marine-derived agents are now in clinical trials for indications such as cancer, anti-inflammatory use and pain. One class of these agents are bryostatin-like compounds, under investigation as anti-cancer therapy.
Chemical diversity
As above mentioned, combinatorial chemistry was a key technology enabling the efficient generation of large screening libraries for the needs of high-throughput screening. However, now, after two decades of combinatorial chemistry, it has been pointed out that despite the increased efficiency in chemical synthesis, no increase in lead or drug candidates has been reached.[55] This has led to analysis of chemical characteristics of combinatorial chemistry products, compared to existing drugs or natural products. The chemoinformatics concept chemical diversity, depicted as distribution of compounds in the chemical space based on their physicochemical characteristics, is often used to describe the difference between the combinatorial chemistry libraries and natural products. The synthetic, combinatorial library compounds seem to cover only a limited and quite uniform chemical space, whereas existing drugs and particularly natural products, exhibit much greater chemical diversity, distributing more evenly to the chemical space.[54] The most prominent differences between natural products and compounds in combinatorial chemistry libraries is the number of chiral centers (much higher in natural compounds), structure rigidity (higher in natural compounds) and number of aromatic moieties (higher in combinatorial chemistry libraries). Other chemical differences between these two groups include the nature of heteroatoms (O and N enriched in natural products, and S and halogen atoms more often present in synthetic compounds), as well as level of non-aromatic unsaturation (higher in natural products). As both structure rigidity and chirality are well-established factors in medicinal chemistry known to enhance compounds specificity and efficacy as a drug, it has been suggested that natural products compare favourably to today's combinatorial chemistry libraries as potential lead molecules.
Screening
Two main approaches exist for the finding of new bioactive chemical entities from natural sources.
The first is sometimes referred to as random collection and screening of material, but the collection is far from random. Biological (often botanical) knowledge is often used to identify families that show promise. This approach is effective because only a small part of the earth's biodiversity has ever been tested for pharmaceutical activity. Also, organisms living in a species-rich environment need to evolve defensive and competitive mechanisms to survive. Those mechanisms might be exploited in the development of beneficial drugs.
A collection of plant, animal and microbial samples from rich ecosystems can potentially give rise to novel biological activities worth exploiting in the drug development process. One example of successful use of this strategy is the screening for antitumor agents by the National Cancer Institute, which started in the 1960s. Paclitaxel was identified from Pacific yew tree Taxus brevifolia. Paclitaxel showed anti-tumour activity by a previously undescribed mechanism (stabilization of microtubules) and is now approved for clinical use for the treatment of lung, breast, and ovarian cancer, as well as for Kaposi's sarcoma. Early in the 21st century, Cabazitaxel (made by Sanofi, a French firm), another relative of taxol has been shown effective against prostate cancer, also because it works by preventing the formation of microtubules, which pull the chromosomes apart in dividing cells (such as cancer cells). Other examples are: 1. Camptotheca (Camptothecin · Topotecan · Irinotecan · Rubitecan · Belotecan); 2. Podophyllum (Etoposide · Teniposide); 3a. Anthracyclines (Aclarubicin · Daunorubicin · Doxorubicin · Epirubicin · Idarubicin · Amrubicin · Pirarubicin · Valrubicin · Zorubicin); 3b. Anthracenediones (Mitoxantrone · Pixantrone).
The second main approach involves ethnobotany, the study of the general use of plants in society, and ethnopharmacology, an area inside ethnobotany, which is focused specifically on medicinal uses.
Artemisinin, an antimalarial agent from sweet wormtree Artemisia annua, used in Chinese medicine since 200BC is one drug used as part of combination therapy for multiresistant Plasmodium falciparum.
Structural elucidation
The elucidation of the chemical structure is critical to avoid the re-discovery of a chemical agent that is already known for its structure and chemical activity. Mass spectrometry is a method in which individual compounds are identified based on their mass/charge ratio, after ionization. Chemical compounds exist in nature as mixtures, so the combination of liquid chromatography and mass spectrometry (LC-MS) is often used to separate the individual chemicals. Databases of mass spectras for known compounds are available and can be used to assign a structure to an unknown mass spectrum. Nuclear magnetic resonance spectroscopy is the primary technique for determining chemical structures of natural products. NMR yields information about individual hydrogen and carbon atoms in the structure, allowing detailed reconstruction of the molecule's architecture.
New Drug Application
When a drug is developed with evidence throughout its history of research to show it is safe and effective for the intended use in the United States, the company can file an application – the New Drug Application (NDA) – to have the drug commercialized and available for clinical application.[75] NDA status enables the FDA to examine all submitted data on the drug to reach a decision on whether to approve or not approve the drug candidate based on its safety, specificity of effect, and efficacy of doses.[75]
See also
- Antitarget
- Bioinformatics
- Biomedical informatics
- Cheminformatics
- Drug discovery hit to lead
- Drug metabolism
- Fragment-based drug discovery
- High content screening
- Pharmacogenetics
- Pharmacognosy
- Physiologically-based pharmacokinetic modelling
- Pre-clinical development
- Protein-directed dynamic combinatorial chemistry
- Discovery and development of proton pump inhibitors
- Discovery and development of melatonin receptor agonists
- Discovery and development of nucleoside and nucleotide reverse transcriptase inhibitors
- Discovery and development of Bcr-Abl tyrosine kinase inhibitors
- Discovery and development of antiandrogens
- Discovery and development of cephalosporins
- Retrometabolic drug design
References
- "The drug development process". US Food and Drug Administration. 4 January 2018. Retrieved 18 December 2019.
- "The drug development process: Step 1: Discovery and development". US Food and Drug Administration. 4 January 2018. Retrieved 18 December 2019.
- Helleboid S, Haug C, Lamottke K, et al. The Identification of Naturally Occurring Neoruscogenin as a Bioavailable, Potent, and High-Affinity Agonist of the Nuclear Receptor RORα (NR1F1). Journal of Biomolecular Screening. 2014;19(3):399–406. https://doi.org/10.1177/1087057113497095.
- Herrmann, A., Roesner, M., Werner, T. et al. Potent inhibition of HIV replication in primary human cells by novel synthetic polyketides inspired by Aureothin. Sci Rep 10, 1326 (2020). https://doi.org/10.1038/s41598-020-57843-9.
- "The drug development process: Step 3: Clinical research". US Food and Drug Administration. 4 January 2018. Retrieved 18 December 2019.
- Anson D, Ma J, He JQ (1 May 2009). "Identifying Cardiotoxic Compounds". Genetic Engineering & Biotechnology News. TechNote. Vol. 29, no. 9. Mary Ann Liebert. pp. 34–35. ISSN 1935-472X. OCLC 77706455. Archived from the original on 21 September 2012. Retrieved 25 July 2009.
- Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL (March 2010). "How to improve R&D productivity: the pharmaceutical industry's grand challenge". Nature Reviews. Drug Discovery. 9 (3): 203–14. doi:10.1038/nrd3078. PMID 20168317. S2CID 1299234.
- Current Model for Financing Drug Development: From Concept Through Approval. Institute of Medicine (US), Forum on Drug Discovery, Development, and Translation, National Academies Press, Washington (DC). 2009.
- Warren J (April 2011). "Drug discovery: lessons from evolution". British Journal of Clinical Pharmacology. 71 (4): 497–503. doi:10.1111/j.1365-2125.2010.03854.x. PMC 3080636. PMID 21395642.
- Takenaka T (September 2001). "Classical vs reverse pharmacology in drug discovery". BJU International. 88 (Suppl 2): 7–10, discussion 49–50. doi:10.1111/j.1464-410X.2001.00112.x. PMID 11589663.
- Lee JA, Uhlik MT, Moxham CM, Tomandl D, Sall DJ (May 2012). "Modern phenotypic drug discovery is a viable, neoclassic pharma strategy". Journal of Medicinal Chemistry. 55 (10): 4527–38. doi:10.1021/jm201649s. PMID 22409666.
- Elion GB (1993). "The quest for a cure". Annual Review of Pharmacology and Toxicology. 33: 1–23. doi:10.1146/annurev.pa.33.040193.000245. PMID 8494337.
- Elion GB. "The purine path to chemotherapy. Nobel Lecture 1988".
- Black J. "Drugs from emasculated hormones: the principles of synoptic antagonism. Nobel Lecture 1988". Retrieved 28 February 2014.
- Endo A. "The discovery of the statins and their development". Retrieved 28 February 2014.
- Watts G (2012). "Obituary: Sir David Jack". The Lancet. 379 (9811): 116. doi:10.1016/S0140-6736(12)60053-1. S2CID 54305535.
- Swinney DC, Anthony J (July 2011). "How were new medicines discovered?". Nature Reviews. Drug Discovery. 10 (7): 507–19. doi:10.1038/nrd3480. PMID 21701501. S2CID 19171881.
- Rask-Andersen M, Almén MS, Schiöth HB (August 2011). "Trends in the exploitation of novel drug targets". Nature Reviews. Drug Discovery. 10 (8): 579–90. doi:10.1038/nrd3478. PMID 21804595. S2CID 3328752.
- Jacobson KA (December 2015). "New paradigms in GPCR drug discovery". Biochemical Pharmacology. 98 (4): 541–555. doi:10.1016/j.bcp.2015.08.085. PMC 4967540. PMID 26265138.
- Chen, Ya; Kirchmair, Johannes (6 September 2020). "Cheminformatics in natural product‐based drug discovery". Molecular Informatics. 39 (12): 2000171. doi:10.1002/minf.202000171. ISSN 1868-1743. PMC 7757247. PMID 32725781.
- Baker M (9 January 2017). "Deceptive curcumin offers cautionary tale for chemists". Nature. 541 (7636): 144–145. doi:10.1038/541144a. PMID 28079090.
- Dahlin JL, Walters MA (July 2014). "The essential roles of chemistry in high-throughput screening triage". Future Medicinal Chemistry. 6 (11): 1265–90. doi:10.4155/fmc.14.60. PMC 4465542. PMID 25163000.
- Baell JB, Holloway GA (April 2010). "New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays". Journal of Medicinal Chemistry. 53 (7): 2719–40. CiteSeerX 10.1.1.394.9155. doi:10.1021/jm901137j. PMID 20131845.
- Hopkins AL, Groom CR, Alex A (May 2004). "Ligand efficiency: a useful metric for lead selection". Drug Discovery Today. 9 (10): 430–1. doi:10.1016/S1359-6446(04)03069-7. PMID 15109945.
- Ryckmans T, Edwards MP, Horne VA, Correia AM, Owen DR, Thompson LR, Tran I, Tutt MF, Young T (August 2009). "Rapid assessment of a novel series of selective CB(2) agonists using parallel synthesis protocols: A Lipophilic Efficiency (LipE) analysis". Bioorganic & Medicinal Chemistry Letters. 19 (15): 4406–9. doi:10.1016/j.bmcl.2009.05.062. PMID 19500981.
- Leeson PD, Springthorpe B (November 2007). "The influence of drug-like concepts on decision-making in medicinal chemistry". Nature Reviews. Drug Discovery. 6 (11): 881–90. doi:10.1038/nrd2445. PMID 17971784. S2CID 205476574.
- Rester U (July 2008). "From virtuality to reality – Virtual screening in lead discovery and lead optimization: a medicinal chemistry perspective". Current Opinion in Drug Discovery & Development. 11 (4): 559–68. PMID 18600572.
- Rollinger JM, Stuppner H, Langer T (2008). "Virtual screening for the discovery of bioactive natural products". Natural Compounds as Drugs Volume I. Progress in Drug Research. Vol. 65. pp. 211, 213–49. doi:10.1007/978-3-7643-8117-2_6. ISBN 978-3-7643-8098-4. PMC 7124045. PMID 18084917.
- Barcellos GB, Pauli I, Caceres RA, Timmers LF, Dias R, de Azevedo WF (December 2008). "Molecular modeling as a tool for drug discovery". Current Drug Targets. 9 (12): 1084–91. doi:10.2174/138945008786949388. PMID 19128219.
- Durrant JD, McCammon JA (October 2011). "Molecular dynamics simulations and drug discovery". BMC Biology. 9: 71. doi:10.1186/1741-7007-9-71. PMC 3203851. PMID 22035460.
- Borhani DW, Shaw DE (January 2012). "The future of molecular dynamics simulations in drug discovery". Journal of Computer-Aided Molecular Design. 26 (1): 15–26. doi:10.1007/s10822-011-9517-y. PMC 3268975. PMID 22183577.
- Ciemny M, Kurcinski M, Kamel K, Kolinski A, Alam N, Schueler-Furman O, Kmiecik S (May 2018). "Protein-peptide docking: opportunities and challenges". Drug Discovery Today. 23 (8): 1530–1537. doi:10.1016/j.drudis.2018.05.006. PMID 29733895.
- Erlanson DA, McDowell RS, O'Brien T (July 2004). "Fragment-based drug discovery". Journal of Medicinal Chemistry. 47 (14): 3463–82. doi:10.1021/jm040031v. PMID 15214773.
- Folkers G, Jahnke W, Erlanson DA, Mannhold R, Kubinyi H (2006). Fragment-based Approaches in Drug Discovery (Methods and Principles in Medicinal Chemistry). Weinheim: Wiley-VCH. ISBN 978-3-527-31291-7.
- Erlanson DA (June 2011). "Introduction to fragment-based drug discovery". Fragment-Based Drug Discovery and X-Ray Crystallography. Topics in Current Chemistry. Vol. 317. pp. 1–32. doi:10.1007/128_2011_180. ISBN 978-3-642-27539-5. PMID 21695633.
- Edward Z, Michael S (2008). Fragment-based drug discovery a practical approach. Wiley.
- Greaney MF, Bhat VT (2010). "Chapter 2: Protein-directed dynamic combinatorial chemistry". In Miller BL (ed.). Dynamic combinatorial chemistry: in drug discovery, bioinorganic chemistry, and materials sciences. New Jersey: John Wiley & Sons. pp. 43–82.
- Huang R, Leung IK (July 2016). "Protein-directed dynamic combinatorial chemistry: a guide to protein ligand and inhibitor discovery". Molecules. 21 (7): 910. doi:10.3390/molecules21070910. PMC 6273345. PMID 27438816.
- Mondal M, Hirsch AK (April 2015). "Dynamic combinatorial chemistry: a tool to facilitate the identification of inhibitors for protein targets". Chemical Society Reviews. 44 (8): 2455–88. doi:10.1039/c4cs00493k. PMID 25706945.
- Herrmann A (March 2014). "Dynamic combinatorial/covalent chemistry: a tool to read, generate and modulate the bioactivity of compounds and compound mixtures". Chemical Society Reviews. 43 (6): 1899–933. doi:10.1039/c3cs60336a. PMID 24296754.
- Hochgürtel M, Lehn JM (2006). "Chapter 16: Dynamic combinatorial diversity in drug discovery". In Jahnke W, Erlanson DA (eds.). Fragment-based approaches in drug discovery. Weinheim: Wiley-VCH. pp. 341–364.
- Caliandro R, Belviso DB, Aresta BM, de Candia M, Altomare CD (June 2013). "Protein crystallography and fragment-based drug design". Future Medicinal Chemistry. 5 (10): 1121–40. doi:10.4155/fmc.13.84. PMID 23795969.
- Chilingaryan Z, Yin Z, Oakley AJ (October 2012). "Fragment-based screening by protein crystallography: successes and pitfalls". International Journal of Molecular Sciences. 13 (10): 12857–79. doi:10.3390/ijms131012857. PMC 3497300. PMID 23202926.
- Valade A, Urban D, Beau JM (January–February 2007). "Two galactosyltransferases' selection of different binders from the same uridine-based dynamic combinatorial library". Journal of Combinatorial Chemistry. 9 (1): 1–4. doi:10.1021/cc060033w. PMID 17206823.
- Zheng W, Thorne N, McKew JC (November 2013). "Phenotypic screens as a renewed approach for drug discovery". Drug Discovery Today. 18 (21–22): 1067–1073. doi:10.1016/j.drudis.2013.07.001. PMC 4531371. PMID 23850704.
- Swinney DC, Anthony J (June 2011). "How were new medicines discovered?". Nature Reviews. Drug Discovery. 10 (7): 507–519. doi:10.1038/nrd3480. PMID 21701501. S2CID 19171881.
- Brown DG, Wobst HJ (March 2020). "Opportunities and Challenges in Phenotypic Screening for Neurodegenerative Disease Research". Journal of Medicinal Chemistry. 63 (5): 1823–1840. doi:10.1021/acs.jmedchem.9b00797. PMID 31268707.
- Moellering RE, Cravatt BF (January 2012). "How chemoproteomics can enable drug discovery and development". Chemistry & Biology. 19 (1): 11–22. doi:10.1016/j.chembiol.2012.01.001. PMC 3312051. PMID 22284350.
- Srinivasan, Bharath (March 2022). "A guide to enzyme kinetics in early drug discovery". The FEBS Journal. doi:10.1111/febs.16404. ISSN 1742-464X.
- Marshall SF, Burghaus R, Cosson V, Cheung SY, Chenel M, DellaPasqua O, et al. (March 2016). "Good Practices in Model-Informed Drug Discovery and Development: Practice, Application, and Documentation". CPT. 5 (3): 93–122. doi:10.1002/psp4.12049. PMC 4809625. PMID 27069774.
- Marshall S, Madabushi R, Manolis E, Krudys K, Staab A, Dykstra K, Visser SA (February 2019). "Model-Informed Drug Discovery and Development: Current Industry Good Practice and Regulatory Expectations and Future Perspectives". CPT. 8 (2): 87–96. doi:10.1002/psp4.12372. PMC 6389350. PMID 30411538.
- Van Wijk RC (2020). "Model-Informed Drug Discovery and Development Strategy for the Rapid Development of Anti-Tuberculosis Drug Combinations". Applied Sciences. 10 (2376): 2376. doi:10.3390/app10072376.
- Reigosa MJ, Pedrol N, González L (2006), Allelopathy: a physiological process with ecological implications, Springer, p. 1, ISBN 978-1-4020-4279-9
- Feher M, Schmidt JM (2003). "Property distributions: differences between drugs, natural products, and molecules from combinatorial chemistry". Journal of Chemical Information and Computer Sciences. 43 (1): 218–27. doi:10.1021/ci0200467. PMID 12546556.
- Newman DJ, Cragg GM (March 2007). "Natural products as sources of new drugs over the last 25 years". Journal of Natural Products. 70 (3): 461–77. doi:10.1021/np068054v. PMID 17309302.
- von Nussbaum F, Brands M, Hinzen B, Weigand S, Häbich D (August 2006). "Antibacterial natural products in medicinal chemistry—exodus or revival?". Angewandte Chemie. 45 (31): 5072–129. doi:10.1002/anie.200600350. PMID 16881035.
The handling of natural products is cumbersome, requiring nonstandardized workflows and extended timelines. Revisiting natural products with modern chemistry and target-finding tools from biology (reversed genomics) is one option for their revival.
- Li JW, Vederas JC (July 2009). "Drug discovery and natural products: end of an era or an endless frontier?". Science. 325 (5937): 161–5. doi:10.1126/science.1168243. PMID 19589993. S2CID 207777087.
With the current framework of HTS in major pharmaceutical industries and increasing government restrictions on drug approvals, it is possible that the number of new natural product–derived drugs could go to zero. However, this is likely to be temporary, as the potential for new discoveries in the longer term is enormous.
- Harvey AL, Edrada-Ebel R, Quinn RJ (2015). "The re-emergence of natural products for drug discovery in the genomics era" (PDF). Nature Reviews. Drug Discovery. 14 (2): 111–29. doi:10.1038/nrd4510. hdl:10072/141449. PMID 25614221. S2CID 12369182.
Here, we review strategies for natural product screening that harness the recent technical advances that have reduced [technical barriers to screening natural products in high-throughput assays]. The growing appreciation of functional assays and phenotypic screens may further contribute to a revival of interest in natural products for drug discovery.
- Newman DJ, Cragg GM (2016). "Natural Products as Sources of New Drugs from 1981 to 2014". Journal of Natural Products. 79 (3): 629–61. doi:10.1021/acs.jnatprod.5b01055. PMID 26852623.
... the utilization of natural products and/or their novel structures, in order to discover and develop the final drug entity, is still alive and well. For example, in the area of cancer, over the time frame from around the 1940s to the end of 2014, of the 175 small molecules approved, 131, or 75%, are other than "S" (synthetic), with 85, or 49%, actually being either natural products or directly derived therefrom.
- Torre BG, Albericio F (2017). "The Pharmaceutical Industry in 2016. An Analysis of FDA Drug Approvals from a Perspective of the Molecule Type". Molecules (Basel, Switzerland). 22 (3): 368. doi:10.3390/molecules22030368. PMC 6155368. PMID 28264468.
The outputs from 2016 indicate the so-called small molecules are losing ground against biologics, biomolecules, and other molecules inspired [by] natural products
- Sutton D (2007). "Pedanios Dioscorides: Recording the Medicinal Uses of Plants". In Huxley R (ed.). The Great Naturalists. London: Thames & Hudson, with the Natural History Museum. pp. 32–37. ISBN 978-0-500-25139-3.
- Ahn K (March 2017). "The worldwide trend of using botanical drugs and strategies for developing global drugs". BMB Reports. 50 (3): 111–116. doi:10.5483/BMBRep.2017.50.3.221. PMC 5422022. PMID 27998396.
- Wink M (September 2015). "Modes of Action of Herbal Medicines and Plant Secondary Metabolites". Medicines. 2 (3): 251–286. doi:10.3390/medicines2030251. PMC 5456217. PMID 28930211.
- Oishi S, Kimura SI, Noguchi S, Kondo M, Kondo Y, Shimokawa Y, Iwao Y, Itai S (January 2018). "New scale-down methodology from commercial to lab scale to optimize plant-derived soft gel capsule formulations on a commercial scale". International Journal of Pharmaceutics. 535 (1–2): 371–378. doi:10.1016/j.ijpharm.2017.11.029. PMID 29154803.
- Fingrut O, Flescher E (April 2002). "Plant stress hormones suppress the proliferation and induce apoptosis in human cancer cells". Leukemia. 16 (4): 608–16. doi:10.1038/sj.leu.2402419. PMID 11960340.
- Zhang M, Zhang MW, Zhang L, Zhang L (24 July 2015). "Methyl jasmonate and its potential in cancer therapy". Plant Signaling & Behavior. 10 (9): e1062199. doi:10.1080/15592324.2015.1062199. PMC 4883903. PMID 26208889.
- Turner JG, Ellis C, Devoto A (2002). "The jasmonate signal pathway". The Plant Cell. 14 (Suppl): S153–64. doi:10.1105/tpc.000679. PMC 151253. PMID 12045275.
- Ahmad P, Rasool S, Gul A, Sheikh SA, Akram NA, Ashraf M, Kazi AM, Gucel S (2016). "Jasmonates: Multifunctional Roles in Stress Tolerance". Frontiers in Plant Science. 7: 813. doi:10.3389/fpls.2016.00813. PMC 4908892. PMID 27379115.
- Rotem R, Heyfets A, Fingrut O, Blickstein D, Shaklai M, Flescher E (March 2005). "Jasmonates: novel anticancer agents acting directly and selectively on human cancer cell mitochondria". Cancer Research. 65 (5): 1984–93. doi:10.1158/0008-5472.CAN-04-3091. PMID 15753398.
- Michelet JF, Olive C, Rieux E, Fagot D, Simonetti L, Galey JB, Dalko-Csiba M, Bernard BA, Pereira R (May 2012). "The anti-ageing potential of a new jasmonic acid derivative (LR2412): in vitro evaluation using reconstructed epidermis Episkin™". Experimental Dermatology. 21 (5): 398–400. doi:10.1111/j.1600-0625.2012.01480.x. PMID 22509841.
- Henriet E, Jäger S, Tran C, Bastien P, Michelet JF, Minondo AM, Formanek F, Dalko-Csiba M, Lortat-Jacob H, Breton L, Vivès RR (September 2017). "A jasmonic acid derivative improves skin healing and induces changes in proteoglycan expression and glycosaminoglycan structure". Biochimica et Biophysica Acta (BBA) - General Subjects. 1861 (9): 2250–2260. doi:10.1016/j.bbagen.2017.06.006. PMID 28602514.
- Klessig DF, Tian M, Choi HW (26 May 2016). "Multiple Targets of Salicylic Acid and Its Derivatives in Plants and Animals". Frontiers in Immunology. 7: 206. doi:10.3389/fimmu.2016.00206. PMC 4880560. PMID 27303403.
- Pierpoint WS (1994). "Salicylic Acid and its Derivatives in Plants: Medicines, Metabolites and Messenger Molecules". In Kuntz M (ed.). Advances in Botanical Research. Vol. 20. pp. 163–235. doi:10.1016/S0065-2296(08)60217-7. ISBN 978-0-12-809447-1.
- Faulkner DJ, Newman DJ, Cragg GM (February 2004). "Investigations of the marine flora and fauna of the Islands of Palau". Natural Product Reports. 21 (1): 50–76. doi:10.1039/b300664f. PMID 15039835.
- "The drug development process. Step 4: FDA drug review". US Food and Drug Administration. 4 January 2018. Retrieved 18 December 2019.
Further reading
- Gad SC (2005). Drug Discovery Handbook. Hoboken, N.J: Wiley-Interscience/J. Wiley. ISBN 978-0-471-21384-0.
- Madsen U, Krogsgaard-Larsen P, Liljefors T (2002). Textbook of Drug Design and Discovery. Washington, DC: Taylor & Francis. ISBN 978-0-415-28288-8.
- Rasmussen N (2014). Gene Jockeys: Life Science and the rise of Biotech Enterprise. Baltimore: Johns Hopkins University Press. ISBN 978-1-42141-340-2.
External links
Drug Discovery at Curlie
| General | |
|---|---|
| Pharmaceutical sciences |
|
| Professions | |
| Practice areas |
|
| Pharmaceutical industry |
|
| |