Fetal hemoglobin
Fetal hemoglobin, or foetal haemoglobin (also hemoglobin F, HbF, or α2γ2) is the main oxygen carrier protein in the human fetus. Hemoglobin F is found in fetal red blood cells, and is involved in transporting oxygen from the mother's bloodstream to organs and tissues in the fetus. It is produced at around 6 weeks of pregnancy [1] and the levels remain high after birth until the baby is roughly 2–4 months old.[2] Hemoglobin F has a different composition from the adult forms of hemoglobin, which allows it to bind (or attach to) oxygen more strongly. This way, the developing fetus is able to retrieve oxygen from the mother's bloodstream, which occurs through the placenta found in the mother's uterus.[3]
| Fetal hemoglobin | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (4 subunits, α2γ2) | ||||||||||||||||
.png.webp) Structure of fetal hemoglobin (HbF). The 2α and 2γ subunits are in red and yellow, respectively, and the iron-containing heme groups in green. From PDB: 4MQJ, by authors Soman, J. and Olson J.S. | ||||||||||||||||
| Protein type | metalloprotein, globulin | |||||||||||||||
| Function | oxygen-transport | |||||||||||||||
| Cofactor(s) | heme (4) | |||||||||||||||
| ||||||||||||||||
In the newborn, levels of hemoglobin F gradually decrease and reach adult levels (less than 1% of total hemoglobin) usually within the first year, as adult forms of hemoglobin begin to be produced.[4] Diseases such as beta thalassemias, which affect components of the adult hemoglobin, can delay this process, and cause hemoglobin F levels to be higher than normal.[5] In sickle cell anemia, increasing the production of hemoglobin F has been used as a treatment to relieve some of the symptoms.[6]
Structure and genetics
Hemoglobin F, like adult hemoglobin (hemoglobin A and hemoglobin A2), has four subunits or chains. Each subunit contains a heme group with an iron element which is key in allowing the binding and unbinding of oxygen. As such, hemoglobin F can adopt two states: oxyhemoglobin (bound to oxygen) and deoxyhemoglobin (without oxygen). As hemoglobin F has 4 heme groups, it can bind to up to four oxygen molecules.[7] It is composed of two α (alpha) subunits and two γ (gamma) subunits, whereas hemoglobin A (97% of total hemoglobin in adults) is composed of two α and two β (beta) subunits.
In humans, the α subunit is encoded on chromosome 16 and the γ subunit is encoded on chromosome 11. There are two very similar genes that code for the α subunit, HBA1 and HBA2. The protein that they produce is identical, but they differ in gene regulatory regions that determine when or how much of the protein is produced. This leads to HBA1 and HBA2 each contributing with 40% and 60%, respectively, of the total of α subunits produced. As a consequence, mutations on the HBA2 gene are expected to have a stronger effect than mutations on the HBA1 gene.[8] There are also two similar copies of the gene coding for the γ subunit, HBG1 and HBG2, but the protein produced is slightly different, just in one protein unit: HBG1 codes for the protein form with an alanine at position 136, whereas HBG2 codes for a glycine (see Archived 2020-07-31 at the Wayback Machine). BCL11A and ZBTB7A are major repressor proteins of hemoglobin F production, by binding to the gene coding for the γ subunit at their promoter region.[9] This happens naturally as the newborn baby starts to switch from producing hemoglobin F to producing hemoglobin A. Some genetic diseases can take place due to mutations to genes coding for components of hemoglobin F. Mutations to HBA1 and HBA2 genes can cause alpha-thalassemia[10] and mutations to the promoter regions of HBG1 and HBG2 can cause hemoglobin F to still be produced after the switch to hemoglobin A should have occurred, which is called hereditary persistence of fetal hemoglobin.[9]
Production
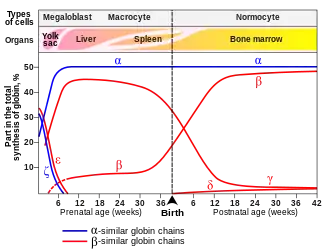
During the first 3 months of pregnancy, the main form of hemoglobin in the embryo/fetus is embryonic hemoglobin, which has 3 variants depending on the types of subunits it contains. The production of hemoglobin F starts from week 6, but it's only from 3 months onwards that it becomes the main type found in fetal red blood cells.[4] The switch to produce adult forms of hemoglobin (essentially hemoglobin A) starts at around 40 weeks of gestation, which is close to the expected time of birth.[1] At birth, hemoglobin F accounts for 50-95% of the infant's hemoglobin and at around 6 months after birth, hemoglobin A becomes the predominant type. By the time the baby is one year old, the proportions of different types of hemoglobin are expected to approximate the adult levels, with hemoglobin F reduced to very low levels.[4] The small proportion of red blood cells containing hemoglobin F are called F-cells, which also contain other types of hemoglobin.
In healthy adults, the composition of hemoglobin is hemoglobin A (~97%), hemoglobin A2 (2.2 - 3.5%) and hemoglobin F (<1%).[11]
Certain genetic abnormalities can cause the switch to adult hemoglobin synthesis to fail, resulting in a condition known as hereditary persistence of fetal hemoglobin.
Binding to oxygen
Factors affecting oxygen affinity
The four hemes, which are the oxygen-binding parts of hemoglobin, are similar between hemoglobin F and other types of hemoglobin, including hemoglobin A. Thus, the key feature that allows hemoglobin F to bind more strongly to oxygen is by having γ subunits (instead of β, for example). In fact, some naturally existing molecules in our body can bind to hemoglobin and change its binding affinity for oxygen. One of the molecules is 2,3-bisphosphoglycerate (2,3-BPG) and it enhances hemoglobin's ability to release oxygen.[12] 2,3-BPG interacts much more with hemoglobin A than hemoglobin F. This is because the adult β subunit has more positive charges than the fetal γ subunit, which attract the negative charges from 2,3-BPG. Due to the preference of 2,3-BPG for hemoglobin A, hemoglobin F binds to oxygen with more affinity, in average.[13]
Even higher oxygen affinity – hemoglobin Barts (four γ subunits)
Hemoglobin Barts is an abnormal form of hemoglobin produced in hemoglobin Barts syndrome or alpha-thalassemia major, the most severe form of alpha-thalassemia. Alpha-thalassemia is a genetic blood disorder and one of the most common hemoglobin-related diseases, affecting the production of α subunits from hemoglobin.[14] Depending on how many genes coding for the α subunit are impacted (between one and four), patients with this disease can have reduced to no production of the α subunit of the hemoglobin. As a consequence, less hemoglobin is available and this affects oxygen supply to the tissues. Hemoglobin Barts syndrome manifests when all four genes coding for α subunit are deleted. This is often fatal for the fetus carrying the disorder, as in the absence of α subunits, a form of hemoglobin with four γ subunits, hemoglobin Barts, is produced. This form of hemoglobin isn't fit for oxygen exchange precisely due to its very high affinity for oxygen. While hemoglobin Barts is very efficient at binding oxygen, it doesn't release oxygen to the organs and tissues.[15] The disease is fatal for the fetus or newborn unless early diagnosis and intervention is carried out during pregnancy, and the child will be dependent on lifelong blood transfusions.
Quantification of oxygen binding
To quantify how strongly a certain type of hemoglobin binds to oxygen (or its affinity for oxygen), a parameter called P50 is often used. In a given situation, P50 can be understood as the partial pressure of oxygen at which Hb is 50% saturated.[16] For example, Hemoglobin F has a lower P50 than hemoglobin A. This means that if we have the same amount of hemoglobin F and hemoglobin A in the blood and add oxygen to it, half of hemoglobin F will bind to oxygen before half of hemoglobin A manages to do so. Therefore, a lower P50 means stronger binding or higher affinity for oxygen.
For reference, the P50 of fetal hemoglobin is roughly 19 mmHg (a measure of pressure), whereas adult hemoglobin is approximately 26.8 mmHg (see Blood gas tension).[17]
Oxygen exchange in the womb
During pregnancy, the mother's circulatory system delivers oxygen and nutrients to the fetus and carries away nutrient-depleted blood enriched with carbon dioxide. The maternal and fetal blood circulations are separate and the exchange of molecules occurs through the placenta, in a region called intervillous space which is located in between maternal and fetal blood vessels.[3]
Focusing on oxygen exchange, there are 3 important aspects that allow it to pass from the maternal circulation into the fetal circulation. Firstly, the presence of hemoglobin F in the fetus allows a stronger binding to oxygen than maternal hemoglobin (see Factors affecting oxygen affinity). Secondly, the mother's bloodstream is richer in oxygen than that of the fetus, so oxygen naturally flows towards the fetal circulation by diffusion.[18] The final factor is related to the effects of pH on maternal and fetal hemoglobin. As the maternal blood acquires more carbon dioxide, it becomes more acidic and this favors the release of oxygen by the maternal hemoglobin. At the same time, the decrease in carbon dioxide in fetal blood makes it more alkaline and favors the uptake of oxygen. This is called the Bohr effect or Haldane effect, which also happens in the air exchange in the lungs.[19] All of these 3 factors are present simultaneously and cooperate to improve the fetus’ access to oxygen from the mother.
F-cells
F-cells are the subpopulation of red blood cells that contain hemoglobin F, in amongst other types of hemoglobin. While common in fetuses, in normal adults, only around 3-7% of red blood cells contain hemoglobin F.[20] The low percentage of F-cells in adults owes to two factors: very low levels of hemoglobin F being present and its tendency to be produced only in a subset of cells rather than evenly distributed amongst all red blood cells. In fact, there is a positive correlation between the levels of hemoglobin F and number of F-cells, with patients with higher percentages of hemoglobin F also having a higher proportion of F-cells.[21] Despite the correlations between hemoglobin F levels and F-cell numbers, usually they are determined by direct measurements. While the amount of hemoglobin F is calculated using cell lysates, which are fluids with contents of cells that were broken open, F-cell numbers are done by counting intact red blood cells.[20]
Due to the correlation between the amount of hemoglobin F and F-cells, F-cell numbers are higher in some inherited hemoglobin disorders, including beta-thalassemia, sickle cell anemia and hereditary persistence of fetal hemoglobin. Additionally, some acquired conditions can also have higher F-cell numbers, such as acute erythropoietic stress (response to poor oxygenation which includes very rapid synthesis of new red blood cells) [22] and pregnancy.[20] F-cells have similar mass of haemoglobin per cell compared to red blood cells without haemoglobin F, which is measured mean cell haemoglobin values (MCH).[23]
Conditions with high hemoglobin F
During pregnancy
There is a significant increase in hemoglobin F levels during early pregnancy. However, it's not clear whether these levels are stable or decrease as the pregnancy goes on, as different sources reported different results.[24][25] The increase in hemoglobin F then induces a 3 to 7 fold increase in the number of F-cells in pregnant women, which was observed between the 23rd to 31st week of gestation.[26] However, as to the reason of the increase in hemoglobin F levels in pregnant women, there doesn't seem to be conclusive evidence. While an early study suggested that maternal red blood cells switch on hemoglobin F production during pregnancy,[26] more recent literature suggested that the increase in haemoglobin F could be, at least in part, due to fetal red blood cells being transferred to the maternal circulation.[27][20]
Presence of high levels of hemoglobin F in pregnant women can impact the growth of the fetus, as fetal red blood cells struggle to compete for the oxygen from the mother's circulation. This is because instead of competing with hemoglobin A, which has a weaker association to oxygen than hemoglobin F, it becomes a competition between fetal and maternal hemoglobin F which have similar affinities for oxygen. As a result, women with hemoglobin F as >70% of total hemoglobin are much more likely to have fetuses that are small for their gestational age compared women with <70% hemoglobin F (at a rate of 100% compared to 8%, respectively).[28]
Hereditary persistence of fetal hemoglobin (HPFH)
This is a rare benign genetic disease where production of hemoglobin F persists after twelve months of life and into the adulthood. As a result, hemoglobin F is present in a higher number of adult red blood cells than normal.[29] It doesn't present symptoms and is usually discovered when screening for other blood-related diseases. In this condition, the genes coding for the γ subunit (HBG1 and HBG2) are not suppressed shortly before birth. This can happen when a mutation occurs in the promoter region of HBG1 and HBG2, preventing the binding of BCL11A and ZBTB7A proteins. These proteins would normally bind and suppress the production of γ subunits and as they can't bind due to the mutation, γ subunits continue to be produced.[9] There are two types of patients with HPFH: either with one normal copy of the gene and one disease form or with two disease copies. Whereas normal adults have less than 1% of hemoglobin F, patients with only one disease gene have 5-30%. Patients with two disease copies can have hemoglobin F in up to 100% of red blood cells.[30] As other diseases such as sickle cell disease could also cause a higher level of hemoglobin F to be present, it can sometimes be misdiagnosed.[31]
Delta beta-thalassemia
Delta beta-thalassemia is a rare genetic blood disorder in which the production of both δ and β subunits are reduced or absent. In these cases, the production of the γ subunit increases to compensate for the loss of δ and β subunits, resulting in a higher amount of hemoglobin F present in the blood. Normally, people have two sets of genes for producing δ and β subunits. People with only one set of working genes don't get any symptoms and in the rarely reported cases where both sets of genes are affected, the patients only experienced mild symptoms.[32]
Clinical significance
Treatment of sickle-cell disease
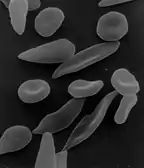
The discovery that hemoglobin F alleviated the symptoms of sickle cell disease occurred in 1948. Janet Watson observed that red blood cells from infants with the disease took longer to sickle and did not deform as much compared to their mother's cells, which carried the disease trait. Later, it was noted that patients with sickle cell trait as well as hereditary persistence of hemoglobin F (HPFH) didn't have symptoms.[33] Additionally, in sickle cell patients, F-cells were found to be more long living than non-F cells as they contain hemoglobin F.
When fetal hemoglobin production is switched off after birth, normal children begin producing adult hemoglobin (HbA). Children with sickle-cell disease begin producing a defective form of hemoglobin called hemoglobin S instead, which form chains that cause red blood cells to change their shape from round to sickle-shaped.[34] These defective red blood cells have a much shorter life span than normal red blood cells (10–20 days compared to up to 120 days).[35] They also have a greater tendency to clump together and block small blood vessels, preventing blood supply to tissues and organs. This leads to the so-called vaso-occlusive crisis, which is a hallmark of the disease.[36] If fetal hemoglobin remains relatively high after birth, the number of painful episodes decreases in patients with sickle-cell disease and they have a better prognosis.[37] Fetal hemoglobin's role in reducing disease severity comes from its ability to disrupt the formation of hemoglobin S chains within red blood cells.[38] Interestingly, while higher levels of hemoglobin F were associated with improvement of some symptoms, including the frequency of painful episodes, leg ulcers and the general severity of the disease, it had no correlation to others. A few examples are priapism, stroke and systemic blood pressure.[33] As hemoglobin F are only produced by some red blood cells, in different quantities, only a subpopulation of cells are protected against sickling. It could be that the symptoms that high hemoglobin F doesn't prevent are quite sensitive to the rupture of the sickled non-F cells.[33]
Hydroxyurea is a chemical that promotes the production of fetal hemoglobin and reduces the premature rupturing of red blood cells.[6][39] Combination therapy with hydroxyurea and recombinant erythropoietin — rather than treatment with hydroxyurea alone — has been shown to further elevate hemoglobin F levels and to promote the development of HbF-containing F-cells.[40]
Hemoglobin F as a marker for cancers
There have been some studies evaluating the possibility of using hemoglobin F as an indicator of the prognosis for cancer. It has been suggested that elevated concentrations of haemoglobin F can be found in main kinds of solid tumours and blood cancers.[41] Examples include acute lymphoblastic leukemia and myeloid leukemia in children, where higher concentrations of hemoglobin F were associated with a worse outcome, including a higher risk of relapse or death.[42] Other cancer types where higher hemoglobin F levels have been observed are transitional cell cancer,[43] colorectal carcinoma[44] and various types of blastomas.[45] In fact, in several types of blastomas, including neuroblastoma and retinoblastoma (affecting the nerve cells and the eyes, respectively), F-cells were found in newly formed blood vessels and spaces in between tumour cells. Clusters of F-cells were also present in the bone marrow of some of these patients.[45] Interestingly, hemoglobin F is not directly produced by tumour cells, but seems to be induced by the biological environment of the cancer in nearby blood cells. A reason suggested for this increase in hemoglobin F is that it may favor cancer growth by providing better oxygen supply to the developing cancerous cells.[43] In adults, increased hemoglobin F production is thought to be caused by factors leading to the activation of the gene coding for the γ subunit, such as DNA demethylation (which can activate normally silent genes and is a hallmark of cancer.[46]
References
- Linch D (1998). Encyclopedia of Immunology (second ed.). Elsevier. ISBN 978-0-12-226765-9.
- Schechter AN (November 2008). "Hemoglobin research and the origins of molecular medicine". Blood. 112 (10): 3927–38. doi:10.1182/blood-2008-04-078188. PMC 2581994. PMID 18988877.
- Wang Y, Zhao S (2010). "Chapter 2: Placental Blood Circulation". Vascular Biology of the Placenta. Morgan & Claypool Life Sciences.
- Wild B (2017). Dacie and Lewis Practical Haematology (12th ed.). Elsevier. ISBN 978-0-7020-6696-2.
- Sripichai O, Fucharoen S (December 2016). "Fetal hemoglobin regulation in β-thalassemia: heterogeneity, modifiers and therapeutic approaches". Expert Review of Hematology. 9 (12): 1129–1137. doi:10.1080/17474086.2016.1255142. PMID 27801605. S2CID 10820279.
- Lanzkron S, Strouse JJ, Wilson R, Beach MC, Haywood C, Park H, et al. (June 2008). "Systematic review: Hydroxyurea for the treatment of adults with sickle cell disease". Annals of Internal Medicine. 148 (12): 939–55. doi:10.7326/0003-4819-148-12-200806170-00221. PMC 3256736. PMID 18458272.
- Costanzo LS (2007). Physiology. Hagerstwon, MD: Lippincott Williams & Wilkins. ISBN 978-0781773119.
- Farashi S, Harteveld CL (May 2018). "Molecular basis of α-thalassemia". Blood Cells, Molecules & Diseases. 70: 43–53. doi:10.1016/j.bcmd.2017.09.004. PMID 29032940.
- Martyn GE, Wienert B, Yang L, Shah M, Norton LJ, Burdach J, et al. (April 2018). "Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding". Nature Genetics. 50 (4): 498–503. doi:10.1038/s41588-018-0085-0. PMID 29610478. S2CID 4690503.
- Karakaş Z, Koç B, Temurhan S, Elgün T, Karaman S, Asker G, et al. (December 2015). "Evaluation of Alpha-Thalassemia Mutations in Cases with Hypochromic Microcytic Anemia: The İstanbul Perspective". Turkish Journal of Haematology. 32 (4): 344–50. doi:10.4274/tjh.2014.0204. PMC 4805326. PMID 26377141.
- Thomas C, Lumb AB (2012). "Physiology of haemoglobin". Continuing Education in Anaesthesia, Critical Care & Pain. 12 (5): 251–256. doi:10.1093/bjaceaccp/mks025.
- Litwack G (2018). "Chapter 8 – Glycolysis and Glocuneogenesis". Human Biochemistry. Academic press. ISBN 978-0-12-383864-3.
- Sears D (2016). "Comparing the molecular structure differences between HbF and HbA that affect BPG binding". Biosci Portal. Retrieved 11 March 2020.
- Galanello R, Cao A (February 2011). "Gene test review. Alpha-thalassemia". Genetics in Medicine. 13 (2): 83–8. doi:10.1097/GIM.0b013e3181fcb468. PMID 21381239.
- Forget BG, Bunn HF (February 2013). "Classification of the disorders of hemoglobin". Cold Spring Harbor Perspectives in Medicine. 3 (2): a011684. doi:10.1101/cshperspect.a011684. PMC 3552344. PMID 23378597.
- Awasthi V, Goins E, Phillips W (2006). "Chapter 43 – Liposome-encapsulated hemoglobin: history, preparation and evaluation". Blood Substitutes. Academic press. ISBN 978-0-12-759760-7.
- Yacov R, Derek K, Namasivayam A (2017). "Chapter 10 – Blood gases: technical aspects and interpretation". Assisted Ventilation of the Neonate (sixth ed.). Elsevier. ISBN 978-0-323-39006-4.
- Metcalfe J, Bartels H, Moll W (October 1967). "Gas exchange in the pregnant uterus". Physiological Reviews. 47 (4): 782–838. doi:10.1152/physrev.1967.47.4.782. PMID 4964061.
- Griffiths S, Campbell J (2015). "Placental structure, function and drug transfer". Continuing Education in Anaesthesia, Critical Care & Pain. 15 (2): 84–89. doi:10.1093/bjaceaccp/mku013.
- Italia KY, Colah R, Mohanty D (December 2007). "Evaluation of F cells in sickle cell disorders by flow cytometry -- comparison with the Kleihauer-Betke's slide method". International Journal of Laboratory Hematology. 29 (6): 409–14. doi:10.1111/j.1365-2257.2006.00884.x. PMID 17988294. S2CID 46171087.
- Wood WG, Stamatoyannopoulos G, Lim G, Nute PE (November 1975). "F-cells in the adult: normal values and levels in individuals with hereditary and acquired elevations of Hb F". Blood. 46 (5): 671–82. doi:10.1182/blood.V46.5.671.bloodjournal465671. PMID 1100141.
- Kim TS, Hanak M, Trampont PC, Braciale TJ (October 2015). "Stress-associated erythropoiesis initiation is regulated by type 1 conventional dendritic cells". The Journal of Clinical Investigation. 125 (10): 3965–80. doi:10.1172/JCI81919. PMC 4607133. PMID 26389678.
- Dover GJ, Boyer SH (April 1987). "Fetal hemoglobin-containing cells have the same mean corpuscular hemoglobin as cells without fetal hemoglobin: a reciprocal relationship between gamma- and beta-globin gene expression in normal subjects and in those with high fetal hemoglobin production". Blood. 69 (4): 1109–13. doi:10.1182/blood.V69.4.1109.bloodjournal6941109. PMID 2435342.
- Ibrahim M, Qari MH, Sait W, Abulela M (2009). "Pattern of HB F level rise during normal pregnancies". Hemoglobin. 33 (6): 534–8. doi:10.3109/03630260903332981. PMID 19958203. S2CID 41124341.
- Yamada T, Morikawa M, Yamada T, Nishida R, Takeda M, Kawaguchi S, Minakami H (January 2013). "Changes in hemoglobin F levels in pregnant women unaffected by clinical fetomaternal hemorrhage". Clinica Chimica Acta; International Journal of Clinical Chemistry. 415: 124–7. doi:10.1016/j.cca.2012.10.002. hdl:2115/53256. PMID 23073220. S2CID 23746089.
- Boyer SH, Belding TK, Margolte L, Noyes AN, Burke PJ, Bell WR (September 1975). "Variations in the frequency of fetal hemoglobin-bearing erythrocytes (F-cells) in well adults, pregnant women, and adult leukemics". The Johns Hopkins Medical Journal. 137 (3): 105–15. PMID 810611.
- Dana M, Fibach E (March 2018). "Fetal Hemoglobin in the Maternal Circulation - Contribution of Fetal Red Blood Cells". Hemoglobin. 42 (2): 138–140. doi:10.1080/03630269.2018.1466712. PMID 29745271. S2CID 13661613.
- Murji A, Sobel ML, Hasan L, McLeod A, Waye JS, Sermer M, Berger H (February 2012). "Pregnancy outcomes in women with elevated levels of fetal hemoglobin". The Journal of Maternal-Fetal & Neonatal Medicine. 25 (2): 125–9. doi:10.3109/14767058.2011.564241. PMID 21473677. S2CID 5500015.
- Hemosh A (9 September 2014). "FETAL HEMOGLOBIN QUANTITATIVE TRAIT LOCUS 1; HBFQTL1". OMIM. Johns Hopkins University. Retrieved 15 March 2020.
- Thein SL, Craig JE (1998). "Genetics of Hb F/F cell variance in adults and heterocellular hereditary persistence of fetal hemoglobin". Hemoglobin. 22 (5–6): 401–14. doi:10.3109/03630269809071538. PMID 9859924.
- Shaukat I, Pudal A, Yassin S, Höti N, Mustafa S (2018). "Blessing in disguise; a case of Hereditary Persistence of Fetal Hemoglobin". Journal of Community Hospital Internal Medicine Perspectives. 8 (6): 380–381. doi:10.1080/20009666.2018.1536241. PMC 6292363. PMID 30559951.
- Wahed A, Dasgupta A (2015). "Chapter 4 – Hemoglobinopathes and Thalassemias". Hematology and Coagulation. Elsevier. ISBN 978-0-12-800241-4.
- Akinsheye I, Alsultan A, Solovieff N, Ngo D, Baldwin CT, Sebastiani P, et al. (July 2011). "Fetal hemoglobin in sickle cell anemia". Blood. 118 (1): 19–27. doi:10.1182/blood-2011-03-325258. PMC 3139383. PMID 21490337.
- "Sickle cell disease". U.S. National Library of Medicine. NIH. 2020-03-15. Retrieved 2020-03-15.
- "Sickle Cell Disease". Johns Hopkins Medicine. The Johns Hopkins University, The Johns Hopkins Hospital, and Johns Hopkins Health System. 2020. Retrieved 16 April 2020.
- Manwani D, Frenette PS (December 2013). "Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies". Blood. 122 (24): 3892–8. doi:10.1182/blood-2013-05-498311. PMC 3854110. PMID 24052549.
- Akinsheye I, Solovieff N, Ngo D, Malek A, Sebastiani P, Steinberg MH, Chui DH (February 2012). "Fetal hemoglobin in sickle cell anemia: molecular characterization of the unusually high fetal hemoglobin phenotype in African Americans". American Journal of Hematology. 87 (2): 217–9. doi:10.1002/ajh.22221. PMC 3302931. PMID 22139998.
- Ma Q, Wyszynski DF, Farrell JJ, Kutlar A, Farrer LA, Baldwin CT, Steinberg MH (December 2007). "Fetal hemoglobin in sickle cell anemia: genetic determinants of response to hydroxyurea". The Pharmacogenomics Journal. 7 (6): 386–94. doi:10.1038/sj.tpj.6500433. PMID 17299377.
- Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, et al. (May 1995). "Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia". The New England Journal of Medicine. 332 (20): 1317–22. doi:10.1056/NEJM199505183322001. PMID 7715639.
- Rodgers GP, Dover GJ, Uyesaka N, Noguchi CT, Schechter AN, Nienhuis AW (January 1993). "Augmentation by erythropoietin of the fetal-hemoglobin response to hydroxyurea in sickle cell disease". The New England Journal of Medicine. 328 (2): 73–80. doi:10.1056/NEJM199301143280201. PMID 7677965.
- Wolk M, Newland AC, De La Salle B (1999). "Refinement of plasma fetal hemoglobin (HbF) measurements, as related to whole blood HbF, in cancer patients". Journal of Tumor.
- Rautonen J, Siimes MA (July 1990). "Initial blood fetal hemoglobin concentration is elevated and is associated with prognosis in children with acute lymphoid or myeloid leukemia". Blut. 61 (1): 17–20. doi:10.1007/BF01739428. PMID 1696840. S2CID 22096967.
- Wolk M, Martin JE (July 2012). "Fetal haemopoiesis marking low-grade urinary bladder cancer". British Journal of Cancer. 107 (3): 477–81. doi:10.1038/bjc.2012.268. PMC 3405209. PMID 22735903.
- Wolk M, Martin JE, Reinus C (June 2006). "Development of fetal haemoglobin-blood cells (F cells) within colorectal tumour tissues". Journal of Clinical Pathology. 59 (6): 598–602. doi:10.1136/jcp.2005.029934. PMC 1860403. PMID 16469830.
- Wolk M, Martin JE, Nowicki M (August 2007). "Foetal haemoglobin-blood cells (F-cells) as a feature of embryonic tumours (blastomas)". British Journal of Cancer. 97 (3): 412–9. doi:10.1038/sj.bjc.6603867. PMC 2360326. PMID 17595660.
- Cheishvili D, Boureau L, Szyf M (June 2015). "DNA demethylation and invasive cancer: implications for therapeutics". British Journal of Pharmacology. 172 (11): 2705–15. doi:10.1111/bph.12885. PMC 4439869. PMID 25134627.
External links
- Hemoglobinopathies
- Transport across the placenta
- American Sickle Cell Anemia Association
- SCDAA: Break The Sickle Cycle
- Hemoglobin synthesis
- Hemoglobin structure and function (archived February 3, 2002
- Hemoglobin F fact sheet (archived October 29, 2009)
- Fetal hemoglobin (doc file; archived March 30 2003)
- Hydroxyurea in sickle-cell disease (archived December 28, 2014 at )
- Chapter 26 Fetal Hemoglobin Induction; Management of Sickle-Cell Disease 4th Edition 2002 (NIH Publication No. 02-2117)