Hemoglobin A
Hemoglobin A (HbA), also known as adult hemoglobin, hemoglobin A1 or α2β2, is the most common human hemoglobin tetramer, accounting for over 97% of the total red blood cell hemoglobin.[1] Hemoglobin is an oxygen-binding protein, found in erythrocytes, which transports oxygen from the lungs to the tissues.[2] Hemoglobin A is the most common adult form of hemoglobin and exists as a tetramer containing two alpha subunits and two beta subunits (α2β2).[3] Hemoglobin A2 (HbA2) is a less common adult form of hemoglobin and is composed of two alpha and two delta-globin subunits. This hemoglobin makes up 1-3% of hemoglobin in adults.[4]
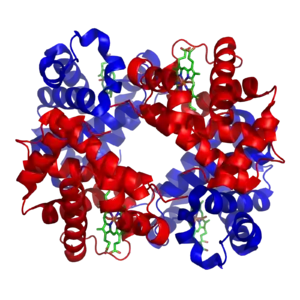
Structure and function
Hemoglobin A (HbA) is the most common adult form of hemoglobin and exists as a tetramer containing two alpha subunits and two beta subunits (α2β2).[3] Each subunit contains a heme group that diatomic oxygen (O2) molecules can bind to.[5] In addition to oxygen, subunit assembly and quaternary structure are known to play important roles in Hb affinity. When hemoglobin binds to O2 (oxyhemoglobin), it will attach to the Iron II (Fe2+) of heme and it is this iron ion that can bind and unbind oxygen to transport oxygen throughout the body.[2] All subunits must be present for hemoglobin to pick up and release oxygen under normal conditions.[6]
Synthesis
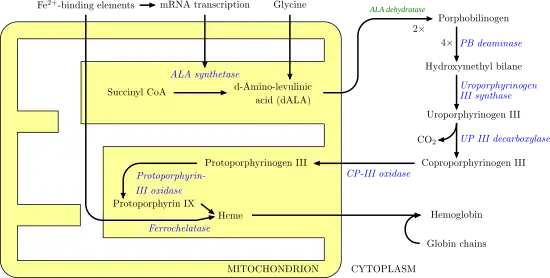
Heme synthesis
Heme synthesis involves a series of enzymatic steps that take place within the mitochondrion and cytosol of the cell. First, in the mitochondrion, the condensation of succinyl CoA and glycine by ALA synthase takes place to produce 5-aminolevulinic acid (ALA). ALA then moves to the cytosol and after a series of reactions creates coproporphyringen III. This molecule moves back into the mitochondrion where it reacts with protoporphyrin-III oxidase to produce protoporphyrin IX. Iron is then enzymatically inserted into protoporphyrin via ferrochelatase to produce heme.[7]
Globin synthesis
Globin synthesis takes place in the ribosomes which are located within the cytosol. Two globin chains that have heme groups combine to form hemoglobin. One of the chains is an alpha chain and the other is a non-alpha chain. Non-alpha chain nature in hemoglobin molecules varies due to different variables. Fetuses have a non-alpha chain called gamma and after birth it is then called beta. The beta chain will pair with the alpha chain. It is the combining of two alpha and non-alpha chains which create a hemoglobin molecule. Two alpha and two gamma chains form fetal hemoglobin or hemoglobin F (HbF). After the first five to six months after birth, the combining of two alpha chains and two beta chains form adult hemoglobin (HbA). The genes that encode for the alpha chains are located on chromosome 16, while the genes that encode for non-alpha chains are located on chromosome 11.[7]
Clinical significance
Due to the numerous steps and processes during hemoglobin synthesis, there are many places in which errors can occur. Heme synthesis involves multiple enzymes and when these enzymes are deficient or do not function properly consequences such as mutations or deletions in genes coding for the globin chain can occur.[2] This results in globin gene disorders (hemoglobinopathies) which can be either abnormal globin chain variants (sickle cell anemia) or reduced chain synthesis in erythroid cells (thalassemia) during the cellular process of hematopoiesis.[8] These hemoglobinopathies are often inherited as autosomal recessive traits.[9]
Alpha-thalassemia
Alpha-thalassemia (α-thalassemia) is defined by a lack of α-globin chain production in hemoglobin, and those who carry a mutation impacting the α-globin chain on only one chromosome are considered to have a “silent” α-thalassemia whereas, if the mutation is on both then it is considered an α-thalassemia trait.[9] α-thalassemia is mostly found in sub-tropical and tropical areas, where individuals who carry the gene is 80-90% of the population.[10] Like other hemoglobin related disorders (sickle cell and β-thalassemia), it is hypothesized that α-thalassemia is selected for within populations due to carriers being better protected against malaria falciparum.[9] Most carriers of α-thalassemia are asymptomatic and are diagnosed if it is found after routine hematological analyses or before birth screenings.[11] Single α-globin gene carriers usually have no profound fatigue or anemia because they have a compensating increase in the number of microcytic red blood cells.[9][11] In contrast, mild α-thalassemia carriers could have symptoms of anemia due to other factors not related specifically to the disorder: poor diet, drop in hemoglobin levels due to blood loss, or other diseases.[9]
The most severe form of α -thalassemia is a condition that begins at infancy in which there is no expression of α-genes and results in a large production of hemoglobin Bart's (Hb Bart's).[11] The most common cause of Hb Bart’s is the inheritance of a deletion allele in that lacks functional α-globin genes from both parents.[9] Hb Bart’s is a tetramer of four gamma-globulin subunits and is ineffective at transporting oxygen to the tissues due to its very high oxygen affinity.[12] This usually results in fatal hydrops fetalis and associated symptoms include intra-uterine anemia, slowing of brain growth, oedema, deformities of the skeleton, and cardiovascular deformities that could lead to cardiac failure.[13]
Beta-thalassemia
Beta-thalassemia (β-thalassemia) is an inherited mutation of the β-globulin gene which causes the reduced synthesis of the β-globin chain of hemoglobin.[14] The majority of the mutations are point mutations that affect translation, transcriptional control, and splicing of the hemoglobin β gene and gene product.[15] Individuals with one gene mutation (heterozygocity) are considered to have β-thalassemia minor (carrier or trait β-thalassemia), while those who have two gene mutations (homozygosity or compound heterozygosity) are diagnosed with β-thalassemia or intermedia.[14][2] Due to the lack of beta-globin, accumulation of alpha-globin subunits and alpha tetramers begin to accumulate leading to damage of erythrocytes.[2] People of Asian, Middle Eastern, and Mediterranean descent have a much higher incidences of β-thalassemia.[14] It has been determined that there is a wide variation in phenotypes and genotypes of the disease due to more than 200 different thalassemia-associated mutations have being found in the beta-globin gene.[15] Individuals with β-thalassemia major usually require medical attention within the first 2 years of life and require regular blood transfusions to survive. Patients who present the disorder later usually do not require transfusions and are diagnosed with thalassemia intermedia.[16]

Sickle cell disease
Sickle hemoglobin (HbS), is the most common variant of hemoglobin and arises due to an amino acid substitution in the beta-globin subunit at the sixth residue from glutamic acid to valine. There are different forms of sickle cell disease. HB SS which is the most common and severe form of sickle cell. Hb SC is due to inheriting Hb S from one parent and Hb C (hemoglobin C) from the other parent. Hb S beta thalassemia is the least common and is experienced in patients who have inherited beta thalassemia hemoglobin from one parent and HbS from the other.[17] In addition, there is sickle cell trait (HbAS) which is defined by having HbA and HbS. This makes the individual heterozygous for sickle cell. Of the world population, it is estimated that there are about 300 million individuals with the sickle cell trait and about 100 million of those are in sub-Saharan Africa.[18] There is also a higher prevalence of sickle cell trait in areas that malaria is commonly found, with the prevalence in some parts of Africa and Saudi Arabia being as high as 25% and 60%, respectively.[19] Individuals who have HbAS have about 40%HbS, 56% HBA, and are usually asymptomatic unless there is a severe lack of oxygen to the body (hypoxia) which can lead to symptoms of sickle cell disease.[12] However, HbAS does not cause vaso-occlusive crisis, which is known to be associated with sickle cell disease.[17]
Patients that are homozygous for HbS have multi-stranded fibers that induce a change in shape of red blood cells from biconcave disks to elongated crescents. The sickling reaction is reversible after re-oxygenating the hemoglobin, therefore, red blood cells can go through cycles of sickling and unsickling depending on the concentration of oxygen present in the bloodstream.[12] Red blood cells that are sickle-shaped lack flexibility and stick to the walls of blood vessels decreasing or stopping the flow of oxygen to nearby tissues. This decrease in oxygen to the tissues cause vaso-occlusive crisis which presents itself in muscle pain and injury to tissues. Some symptoms of sickle cell anemia include fever, fatigue from anemia, swelling of the hands and feet, stroke, and organ failure.[20] Current treatments include blood transfusions which aid with increasing the number of normal red blood cells, bone marrow transplants to help the patients body produce healthy red blood cells, and medications to help alleviate the symptoms listed previously.[21]
See also
Hemoglobin variants:
Hemoglobin protein subunits (genes):
References
- "Hemoglobinopathies". Retrieved 2009-02-06.
- Farid, Yostina; Lecat, Paul (2019), "Biochemistry, Hemoglobin Synthesis", StatPearls, StatPearls Publishing, PMID 30725597, retrieved 2019-04-10
- Immigrant medicine. Walker, Patricia Frye., Barnett, Elizabeth D. (Elizabeth Day). St. Louis, Mo.: Elsevier Mosby. 2007. ISBN 9780323034548. OCLC 489070888.
{{cite book}}: CS1 maint: others (link) - Kato, Gregory J.; Piel, Frédéric B.; Reid, Clarice D.; Gaston, Marilyn H.; Ohene-Frempong, Kwaku; Krishnamurti, Lakshmanan; Smith, Wally R.; Panepinto, Julie A.; Weatherall, David J. (2018-03-15). "Sickle cell disease" (PDF). Nature Reviews Disease Primers. 4: 18010. doi:10.1038/nrdp.2018.10. hdl:10044/1/57817. ISSN 2056-676X. PMID 29542687. S2CID 3870507.
- Barrick, Doug; Lukin, Jonathan A; Simplaceanu, Virgil; Ho, Chien (2004), "Nuclear Magnetic Resonance Spectroscopy in the Study of Hemoglobin Cooperativity", Methods in Enzymology, Elsevier, 379: 28–54, doi:10.1016/s0076-6879(04)79002-3, ISBN 9780121827830, PMID 15051350
- "Hemoglobin Overview". sickle.bwh.harvard.edu. Retrieved 2019-04-10.
- "Hemoglobin Synthesis". sickle.bwh.harvard.edu. Retrieved 2019-04-11.
- Weatherall, D. J. (1980–1981). "The thalassemia syndromes". Texas Reports on Biology and Medicine. 40: 323–333. ISSN 0040-4675. PMID 7034274.
- Farashi, Samaneh; Harteveld, Cornelis L. (May 2018). "Molecular basis of α-thalassemia". Blood Cells, Molecules and Diseases. 70: 43–53. doi:10.1016/j.bcmd.2017.09.004. PMID 29032940.
- Piel, Frédéric B.; Weatherall, David J. (2014-11-13). Longo, Dan L. (ed.). "The α-Thalassemias" (PDF). New England Journal of Medicine. 371 (20): 1908–1916. doi:10.1056/NEJMra1404415. hdl:10044/1/40453. ISSN 0028-4793. PMID 25390741.
- Muncie, Herbert L.; Campbell, James (2009-08-15). "Alpha and beta thalassemia". American Family Physician. 80 (4): 339–344. ISSN 1532-0650. PMID 19678601.
- Forget, B. G.; Bunn, H. F. (2013-02-01). "Classification of the Disorders of Hemoglobin". Cold Spring Harbor Perspectives in Medicine. 3 (2): a011684. doi:10.1101/cshperspect.a011684. ISSN 2157-1422. PMC 3552344. PMID 23378597.
- Somervaille, Tim (November 2001). "Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management". Journal of the Royal Society of Medicine. 94 (11): 602–603. doi:10.1177/014107680109401119. ISSN 0141-0768. PMC 1282256.
- Needs, Todd; Lynch, David T. (2019), "Beta Thalassemia", StatPearls, StatPearls Publishing, PMID 30285376, retrieved 2019-04-10
- Cao, Antonio; Galanello, Renzo (February 2010). "Beta-thalassemia". Genetics in Medicine. 12 (2): 61–76. doi:10.1097/GIM.0b013e3181cd68ed. ISSN 1098-3600. PMID 20098328.
- Ashiotis, Th.; Zachariadis, Z.; Sofroniadou, K.; Loukopoulos, D.; Stamatoyannopoulos, G. (1973-04-07). "Thalassaemia in Cyprus". British Medical Journal. 2 (5857): 38–42. doi:10.1136/bmj.2.5857.38. ISSN 0007-1447. PMC 1588975. PMID 4695698.
- Ashorobi, Damilola; Bhatt, Ruchi (2019), "Sickle Cell Trait", StatPearls, StatPearls Publishing, PMID 30725815, retrieved 2019-04-10
- Tsaras, Geoffrey; Owusu-Ansah, Amma; Boateng, Freda Owusua; Amoateng-Adjepong, Yaw (June 2009). "Complications Associated with Sickle Cell Trait: A Brief Narrative Review". The American Journal of Medicine. 122 (6): 507–512. doi:10.1016/j.amjmed.2008.12.020. PMID 19393983.
- Li, Eileena J.; Carroll, Vanessa G. (September 2014). "Sickle Cell Trait and Renal Papillary Necrosis". Clinical Pediatrics. 53 (10): 1013–1015. doi:10.1177/0009922814533418. ISSN 0009-9228. PMID 24807983. S2CID 13268104.
- "Sickle Cell Disease | National Heart, Lung, and Blood Institute (NHLBI)". www.nhlbi.nih.gov. Retrieved 2019-04-11.
- "Sickle cell anemia - Diagnosis and treatment - Mayo Clinic". www.mayoclinic.org. Retrieved 2019-04-11.
External links
- Hemoglobin+A at the US National Library of Medicine Medical Subject Headings (MeSH)