Fibrosis
Fibrosis, also known as fibrotic scarring, is a pathological wound healing in which connective tissue replaces normal connective tissue, epithelium tissue of any muscilar tissue to the extent that it goes unchecked, leading to considerable tissue remodelling and the formation of permanent scar tissue.[1][2]
| Fibrosis | |
|---|---|
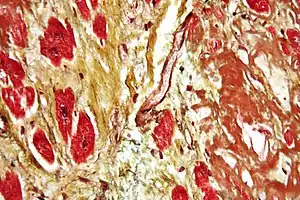 | |
| Micrograph of a heart showing fibrosis (yellow – left of image) and amyloid deposition (brown – right of image). Movat's stain. | |
| Specialty | Pathology, rheumatology |
| Complications | Cirrhosis |
| Risk factors | Repeated injuries, chronic inflammation.[1] |
Repeated injuries, chronic inflammation and repair are susceptible to fibrosis where an accidental excessive accumulation of extracellular matrix components, such as the collagen is produced by fibroblasts, leading to the formation of a permanent fibrotic scar.[1]
In response to injury, this is called scarring, and if fibrosis arises from a single cell line, this is called a fibroma. Physiologically, fibrosis acts to deposit connective tissue, which can interfere with or totally inhibit the normal architecture and function of the underlying organ or tissue. Fibrosis can be used to describe the pathological state of excess deposition of fibrous tissue, as well as the process of connective tissue deposition in healing.[3] Defined by the pathological accumulation of extracellular matrix (ECM) proteins, fibrosis results in scarring and thickening of the affected tissue — it is in essence an exaggerated wound healing response which interferes with normal organ function.[4]
Physiology
Fibrosis is similar to the process of scarring, in that both involve stimulated fibroblasts laying down connective tissue, including collagen and glycosaminoglycans. The process is initiated when immune cells such as macrophages release soluble factors that stimulate fibroblasts. The most well characterized pro-fibrotic mediator is TGF beta, which is released by macrophages as well as any damaged tissue between surfaces called interstitium. Other soluble mediators of fibrosis include CTGF, platelet-derived growth factor (PDGF), and interleukin 10 (IL-10). These initiate signal transduction pathways such as the AKT/mTOR[5] and SMAD[6] pathways that ultimately lead to the proliferation and activation of fibroblasts, which deposit extracellular matrix into the surrounding connective tissue. This process of tissue repair is a complex one, with tight regulation of extracellular matrix (ECM) synthesis and degradation ensuring maintenance of normal tissue architecture. However, the entire process, although necessary, can lead to a progressive irreversible fibrotic response if tissue injury is severe or repetitive, or if the wound healing response itself becomes deregulated.[4][7]
Anatomical location
Fibrosis can occur in many tissues within the body, typically as a result of inflammation or damage, and examples include:
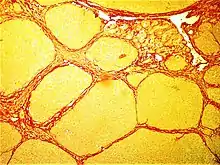
Lungs
- Fibrothorax
- Pulmonary fibrosis
- Cystic fibrosis
- Idiopathic pulmonary fibrosis (idiopathic meaning the cause is unknown)
- Radiation-induced lung injury (following treatment for cancer)
Liver
- Bridging fibrosis An advanced stage of liver fibrosis seen in the progressive form of chronic liver diseases. The term “bridging” means ‘the formation of “bridge” (by the band of mature & thick fibrous tissue) obliterating portal area to central vein’, leads to the formation of pseudolobules. Long-term exposure to hepatotoxin (e.g. thioacetamide, carbon tetrachloride, diethylnitrosamine) results in the bridging fibrosis in experimental animal models.[8]
- Senescence of hepatic stellate cells could prevent progression of liver fibrosis, although this has not been implemented as a therapy, and would carry the risk of hepatic dysfunction.[9]
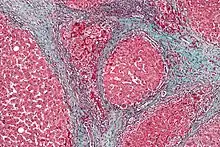
Kidney
- CYR61 induction of cellular senescence in the kidney is a potential therapy to limit fibrosis.[10]
Brain
Heart
Myocardial fibrosis has mainly two forms:
- Interstitial fibrosis, which has been described in congestive heart failure, hypertension, and normal aging.[11]
- Replacement fibrosis, which indicates an older myocardial infarction.[11]
 Healthy myocardium versus interstitial fibrosis in dilated cardiomyopathy. Alcian blue stain.
Healthy myocardium versus interstitial fibrosis in dilated cardiomyopathy. Alcian blue stain. Replacement fibrosis in myocardial infarction, being boundless and dense.
Replacement fibrosis in myocardial infarction, being boundless and dense.
Other
- Arterial stiffness
- Arthrofibrosis (knee, shoulder, other joints)
- Chronic kidney disease[12]
- Crohn's disease (intestine)
- Dupuytren's contracture (hands, fingers)
- Keloid (skin)
- Mediastinal fibrosis (soft tissue of the mediastinum)
- Myelofibrosis (bone marrow)
- Peyronie's disease (penis)
- Nephrogenic systemic fibrosis (skin)
- Progressive massive fibrosis (lungs); a complication of coal workers' pneumoconiosis
- Retroperitoneal fibrosis (soft tissue of the retroperitoneum)
- Scleroderma/systemic sclerosis (skin, lungs)
- Some forms of adhesive capsulitis (shoulder)
References
- Wynn, Thomas A. (2004). "Fibrotic disease and the TH1/TH2 paradigm". Nature Reviews. Immunology. Springer Science and Business Media LLC. 4 (8): 583–594. doi:10.1038/nri1412. ISSN 1474-1733. PMC 2702150. PMID 15286725.
- Birbrair, Alexander; Zhang, Tan; Files, Daniel C.; Mannava, Sandeep; Smith, Thomas; Wang, Zhong-Min; Messi, Maria L.; Mintz, Akiva; Delbono, Osvaldo (2014-11-06). "Type-1 pericytes accumulate after tissue injury and produce collagen in an organ-dependent manner". Stem Cell Research & Therapy. 5 (6): 122. doi:10.1186/scrt512. ISSN 1757-6512. PMC 4445991. PMID 25376879.
- Glossary of dermatopathological terms. DermNet NZ
- Neary R, Watson CJ, Baugh JA (2015). "Epigenetics and the overhealing wound: the role of DNA methylation in fibrosis". Fibrogenesis & Tissue Repair. 8: 18. doi:10.1186/s13069-015-0035-8. PMC 4591063. PMID 26435749.
- Mitra A, Luna JI, Marusina AI, Merleev A, Kundu-Raychaudhuri S, Fiorentino D, Raychaudhuri SP, Maverakis E (2015). "Dual mTOR Inhibition Is Required to Prevent TGF-β-Mediated Fibrosis: Implications for Scleroderma". J Invest Dermatol. 135 (11): 2873–6. doi:10.1038/jid.2015.252. PMC 4640976. PMID 26134944.
- Leask A, Abraham DJ (2004). "TGF-beta signaling and the fibrotic response". FASEB Journal. 18 (7): 816–827. CiteSeerX 10.1.1.314.4027. doi:10.1096/fj.03-1273rev. PMID 15117886. S2CID 2027993.
- Meyer, Keith C. (May 2017). "Pulmonary fibrosis, part I: epidemiology, pathogenesis, and diagnosis". Expert Review of Respiratory Medicine. 11 (5): 343–359. doi:10.1080/17476348.2017.1312346. ISSN 1747-6356. PMID 28345383.
- dwivedi, durgesh Kumar; Jena, Gopa Bandhu (Nov 2018). "Glibenclamide protects against thioacetamide-induced hepatic damage in Wistar rat: investigation on NLRP3, MMP-2, and stellate cell activation". Naunyn-Schmiedeberg's Archives of Pharmacology. 391 (11): 1257–1274. doi:10.1007/s00210-018-1540-2. PMID 30066023. S2CID 51890984.
- Zhang M, Serna-Salas S, Moshage H (2021). "Hepatic stellate cell senescence in liver fibrosis: Characteristics, mechanisms and perspectives". Mechanisms of Ageing and Development. 199: 111572. doi:10.1016/j.mad.2021.111572. PMID 34536446. S2CID 237524296.
- Valentijn FA, Falke LL, Nguyen TQ, Goldschmeding R (2018). "Cellular senescence in the aging and diseased kidney". Journal of Cell Communication and Signaling. 12 (1): 69–82. doi:10.1007/s12079-017-0434-2. PMC 5842195. PMID 29260442.
- Chute, Michael; Aujla, Preetinder; Jana, Sayantan; Kassiri, Zamaneh (2019). "The Non-Fibrillar Side of Fibrosis: Contribution of the Basement Membrane, Proteoglycans, and Glycoproteins to Myocardial Fibrosis". Journal of Cardiovascular Development and Disease. 6 (4): 35. doi:10.3390/jcdd6040035. ISSN 2308-3425. PMC 6956278. PMID 31547598.
- Duffield JS (June 2014). "Cellular and molecular mechanisms in kidney fibrosis". J Clin Invest. 124 (6): 2299–306. doi:10.1172/JCI72267. PMC 4038570. PMID 24892703.
External links
 Media related to Fibrosis at Wikimedia Commons
Media related to Fibrosis at Wikimedia Commons