Galactose epimerase deficiency
Galactose epimerase deficiency, also known as GALE deficiency, Galactosemia III[1] and UDP-galactose-4-epimerase deficiency,[2] is a rare, autosomal recessive form of galactosemia associated with a deficiency of the enzyme galactose epimerase.
| Galactose epimerase deficiency | |
|---|---|
| Other names | Uridine diphosphate galactose-4-epimerase deficiency |
 | |
| Uridine diphosphate glucose | |
Symptoms and signs
Symptoms of congenital Type III Galactosemia are apparent from birth, but vary in severity depending on whether the peripheral or generalized disease form is present. Symptoms may include:[3][4]
- Infantile jaundice
- Infantile hypotonia
- Dysmorphic features
- Sensorineural hearing loss
- Impaired growth
- Cognitive deficiencies
- Depletion of cerebellar Purkinje cells
- Ovarian failure (POI) and hypertrophic hypergonadism
- Liver failure
- Renal failure
- Splenomegaly
- Cataracts
Studies of Type III galactosemia symptoms are mostly descriptive, and precise pathogenic mechanisms remain unknown. This is largely due to a lack of functional animal models of classic galactosemia. The recent development of a Drosophila melanogaster GALE mutant exhibiting galactosemic symptoms may yield a promising future animal model.[3]
Genetics

Galactose epimerase deficiency is an autosomal recessive disorder,[5] which means the defective gene is located on an autosome, and two copies of the defective gene - one from each parent - are required to inherit the disorder. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder.
Genetic basis
Various human GALE mutations resulting in Type III galactosemia have been identified.[6] Functional analysis of these mutant GALE isoforms suggests that reduced catalytic efficiency and increased likelihood of proteolytic digestion act causatively in Type III galactosemia.[6]
| Mutated Residue | Biochemical Effect | Clinical Manifestation |
|---|---|---|
| V94M, K257R, L313M, R335H | Strongly impaired turnover number and specificity constant | Severe generalized galactosemia.[3] |
| S81R, T150M, P293L | Mild turnover number impairment | Intermediate galactosemia.[6] |
| L183P, D103G, G90E, N34S | Strongly impaired turnover number and specificity constant; increased proteolytic digestion. | Severe generalized galactosemia.[3] |
Biochemical basis
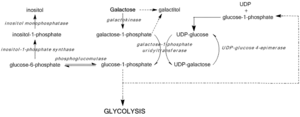
GALE deficiency inhibits UDP-glucose regeneration, preventing the formation of glucose-1-phosphate and leading to the accumulation of galactose and galactose-1-phosphate. High galactose-1-phosphate levels have been shown to interfere with phosphoglucomutase,[7] glycogen phosphorylase,[8] UDP-glycopyrophosphorylase,[9] activity in bacterial models and in vitro, yet in vivo mechanisms toxicity have yet to be confirmed.[3] Regardless, median galactose-1-phosphate levels act as the most accurate predictors of the severity of symptoms associated with Type III galactosemia.[10]
Blockage of the Leloir pathway by GALE deficiency or dysfunction activates alternate pathways of glucose metabolism and leads to galactitol and galactonate formation. Galactonate is metabolized by the pentose phosphate pathway, and is not considered toxic.[11] Galactitol, however, may accumulate in lens fibers, perturbing lens epithelial cell permeability and leading to cell death and cataract formation.[12] GALE deficiency also perturbs glycolipid and glycoprotein biosynthesis due to decreased production of UDP-GalNAc from UDP-GlcNAc.[3]
Diagnosis
Screening for elevated galactose levels may detect GALE deficiency or dysfunction in infants, and mutation studies for GALE are clinically available.[13]
Classification
There are 2 forms of epimerase deficiency: benign RBC deficiency and Severe liver deficiency. Severe form is similar to galactosemia.
Treatment
Individuals presenting with Type III galactosemia must consume a lactose- and galactose-restricted diet devoid of dairy products and mucilaginous plants.[4] Dietary restriction is the only current treatment available for GALE deficiency. As glycoprotein and glycolipid metabolism generate endogenous galactose, however, Type III galactosemia may not be resolved solely through dietary restriction.[3]
References
- Online Mendelian Inheritance in Man (OMIM): Galactose epimerase deficiency - 230350
- Online Mendelian Inheritance in Man (OMIM): UDP-Galactose-4-Epimerase - 606953
- Lai K, Elsas LJ, Wierenga KJ (November 2009). "Galactose toxicity in animals". IUBMB Life. 61 (11): 1063–74. doi:10.1002/iub.262. PMC 2788023. PMID 19859980.
- Walter JH, Roberts RE, Besley GT, Wraith JE, Cleary MA, Holton JB, MacFaul R (April 1999). "Generalised uridine diphosphate galactose-4-epimerase deficiency". Arch. Dis. Child. 80 (4): 374–6. doi:10.1136/adc.80.4.374. PMC 1717903. PMID 10086948.
- Park HD, Park KU, Kim JQ, Shin CH, Yang SW, Lee DH, Song YH, Song J (November 2005). "The molecular basis of UDP-galactose-4-epimerase (GALE) deficiency galactosemia in Korean patientsxz". Genetics in Medicine. 7 (9): 646–9. doi:10.1097/01.gim.0000194023.27802.2d. PMID 16301867.
- Timson DJ (December 2005). "Functional analysis of disease-causing mutations in human UDP-galactose 4-epimerase". FEBS J. 272 (23): 6170–7. doi:10.1111/j.1742-4658.2005.05017.x. PMID 16302980.
- de Jongh WA, Bro C, Ostergaard S, Regenberg B, Olsson L, Nielsen J (October 2008). "The roles of galactitol, galactose-1-phosphate, and phosphoglucomutase in galactose-induced toxicity in Saccharomyces cerevisiae". Biotechnol. Bioeng. 101 (2): 317–26. doi:10.1002/bit.21890. PMID 18421797. S2CID 205497901.
- Maddaiah VT, Madsen NB (September 1966). "Kinetics of purified liver phosphorylase". J. Biol. Chem. 241 (17): 3873–81. PMID 5920799.
- Lai K, Elsas LJ (May 2000). "Overexpression of human UDP-glucose pyrophosphorylase rescues galactose-1-phosphate uridyltransferase-deficient yeast". Biochem. Biophys. Res. Commun. 271 (2): 392–400. doi:10.1006/bbrc.2000.2629. PMID 10799308.
- Guerrero NV, Singh RH, Manatunga A, Berry GT, Steiner RD, Elsas LJ (December 2000). "Risk factors for premature ovarian failure in females with galactosemia". J. Pediatr. 137 (6): 833–41. doi:10.1067/mpd.2000.109148. PMID 11113841.
- Wehrli SL, Berry GT, Palmieri M, Mazur A, Elsas L, Segal S (December 1997). "Urinary galactonate in patients with galactosemia: quantitation by nuclear magnetic resonance spectroscopy". Pediatr. Res. 42 (6): 855–61. doi:10.1203/00006450-199712000-00022. PMID 9396569.
- Kinoshita JH, Dvornik D, Kraml M, Gabbay KH (June 1968). "The effect of an aldose reductase inhibitor on the galactose-exposed rabbit lens". Biochim. Biophys. Acta. 158 (3): 472–5. doi:10.1016/0304-4165(68)90305-x. PMID 5660111.
- Alano A, Almashanu S, Chinsky JM, Costeas P, Blitzer MG, Wulfsberg EA, Cowan TM (June 1998). "Molecular characterization of a unique patient with epimerase-deficiency galactosaemia". J. Inherit. Metab. Dis. 21 (4): 341–50. doi:10.1023/A:1005342306080. PMID 9700591. S2CID 27586949.