Gitelman syndrome
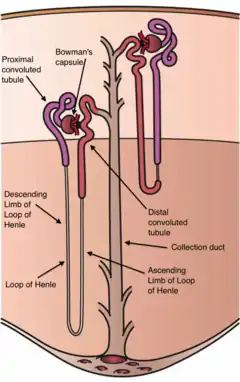
Gitelman syndrome (GS) is an autosomal recessive kidney tubule disorder characterized by low blood levels of potassium and magnesium, decreased excretion of calcium in the urine, and elevated blood pH.[2] The disorder is caused by genetic mutations resulting in improper function of the thiazide-sensitive sodium-chloride symporter (SLC12A3, also known as NCC, NCCT, or TSC) located in the distal convoluted tubule of the kidney.[2] The distal convoluted tubule of the kidney plays an important homoestatic role in sodium and chloride absorption as well as of the reabsorption of magnesium and calcium.[3]
| Gitelman syndrome | |
|---|---|
| Other names | Primary renal tubular hypokalemic hypomagnesemia with hypocalciuria |
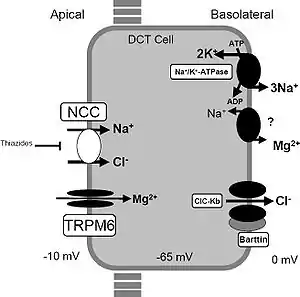 | |
| A model of transport mechanisms in the distal convoluted tubule. Sodium chloride (NaCl) enters the cell via the apical thiazide-sensitive NCC and leaves the cell through the basolateral Cl− channel (ClC-Kb), and the Na+/K+-ATPase. Indicated also are the recently identified magnesium channel TRPM6 in the apical membrane, and a putative Na/Mg exchanger in the basolateral membrane. These transport mechanisms play a role in familial hypokalemia-hypomagnesemia or Gitelman syndrome. | |
| Specialty | Endocrinology |
| Causes | Mutations in SLC12A3, CLCKNB, MT-TI, MT-TF |
Genetic mutations of NCC, lead to loss of function and subsequently, reduced transport of sodium and chloride via NCC. Secondary derangement of calcium, magnesium, and potassium concentrations are caused by secondary effects in the distal tubule and collecting duct. The effect is an electrolyte imbalance similar to that seen with thiazide diuretic therapy (which causes pharmacological inhibition of NCC activity).
Gitelman syndrome was formerly considered a subset of Bartter syndrome until the distinct genetic and molecular bases of these disorders were identified. Bartter syndrome is also an autosomal recessive cause of hypokalemic metabolic alkalosis, but it derives from a mutations of a number of genes that reduce NKCC2 activity. NKCC2 is found in the thick ascending limb of the loop of Henle.[4]
Signs and symptoms
Affected individuals may not have symptoms in some cases.[2] Symptomatic individuals present with symptoms identical to those of patients who are on thiazide diuretics, given that the affected transporter is the exact target of thiazides,[5] (unlike in Bartter syndrome, in which patients present as though on loop diuretics).
Clinical signs of Gitelman syndrome include a high blood pH in combination with low levels of chloride, potassium, and magnesium in the blood and decreased calcium excretion in the urine.[2] In contrast to people with Gordon's syndrome, those affected by Gitelman syndrome generally have low or normal blood pressure. Individuals affected by Gitelman syndrome often complain of severe muscle cramps or weakness, numbness, thirst, waking up at night to urinate, salt cravings, abnormal sensations, chondrocalcinosis, or weakness expressed as extreme fatigue or irritability.[2] Though cravings for salt are most common and severe, cravings for sour foods (e.g. vinegar, lemons, and sour figs) have been noted in some persons affected.[6] More severe symptoms such as seizures, tetany, and paralysis have been reported.[2] Abnormal heart rhythms and a prolonged QT interval can be detected on electrocardiogram[2] and cases of sudden cardiac death have been reported due to low potassium levels. Quality of life is decreased in Gitelman syndrome[7]
Phenotypic variations observed among patients probably result from differences in their genetic background and may depend on which particular amino acid in the NCCT protein has been mutated. A study by Riviera-Munoz et al. identified a subset of individuals with Gitelman syndrome with a severe phenotypic expression. The clinical manifestations observed in this group were neuromuscular manifestations, growth retardation, and ventricular arrhythmias. The patients were mostly male and were found to have at least one allele of a splice defect on the SLC12A3 gene.[8]
Cause
The sodium chloride symporter is a protein made up of 1021 amino acids and 12 transmembrane domains.[9] Mutations that occur on the SLC12A3 gene range from missense, nonsense, frame-shift and splice-site mutations which occur throughout the gene.[9]
Most cases of Gitelman syndrome are linked to inactivating mutations in the SLC12A3 gene, resulting in a loss of function of the thiazide-sensitive sodium-chloride co-transporter (NCCT).[2] This genetic mutation in SLC12A3 is present in 80% of adults with Gitelman syndrome.[2] More than 180 mutations of this transporter protein have been described.[2] This cell membrane protein participates in the control of ion homeostasis at the distal convoluted tubule portion of the nephron. Loss of this transporter also has the indirect effect of increasing calcium reabsorption in a transcellular fashion. This has been suggested to be the result of a putative basolateral Na+/Ca2+ exchanger and apical calcium channel.
When the sodium-chloride cotransporter (NCCT) is inactivated, continued action of the basolateral Na+/K+-ATPase creates a favourable sodium gradient across the basolateral membrane. This increases the reabsorption of divalent cations by secondary active transport. It is currently unknown why calcium reabsorption is increased while magnesium absorption is decreased, often leading to a low level of magnesium in the blood .
A secondary effect of the inactivated sodium-chloride cotransporter is the subsequent activation of the renin-angiotensin aldosterone system (RAAS). RAAS activation is a byproduct of the failure of the distal convoluted tubule in reuptaking electrolytes specifically sodium and chloride leading to cellular dehydration. RAAS attempts to compensate for this dehydration resulting in low serum blood potassium.[10]
A small percentage of Gitelman syndrome cases can be attributed to mutations in the CLCNKB gene. This gene is related to the function of the renal chloride channel CLC-Kb located at the basolateral membrane of cells in the thick ascending limb of the Henle's loop. Genetic variations or mutations in the CLCNKB was initially linked to classic Bartter Syndrome. When mutations are not found within the SLC12A3 gene, screening can be done to rule out involvement of CLCNKB gene.[9]
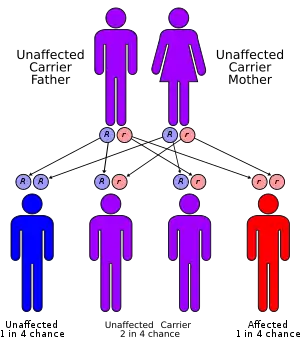
Gitelman syndrome is inherited in an autosomal-recessive manner: one defective allele has to be inherited from each parent.[11]
In 2021, mutations in the tRNAs encoding Isoleucine (MT-TI) and Phenylalanine (MT-TF) in the mitochondrial DNA were described to cause Gitelman syndrome.[12] These homomplasmic mtDNA mutations are maternally inherited.
Diagnosis
Diagnosis of Gitelman syndrome can be confirmed after eliminating other common pathological sources of hypokalemia and metabolic alkalosis.[10] A complete metabolic panel (CMP) or basic metabolic panel (BMP) can be used to evaluate serum electrolyte levels. Renin and aldosterone can be tested in the blood. Electrolyte measurement and aldosterone levels can be done via urine.[10] The pathognomonic clinical markers include low serum levels of potassium, sodium, chloride, and magnesium in the blood as a result of urinary excretion.[13] Urinary fractional excretion potassium is high or inappropriately normal in the context of hypokalaemia, and high levels of urinary sodium and chloride are observed. Other clinical indicators include elevated serum renin and aldosterone in the bloodstream, and metabolic alkalosis. The symptomatic features of this syndrome are highly variable ranging from asymptomatic to mild manifestations (weakness, cramps) to severe symptoms (tetany, paralysis, rhabdomyolysis).[10] Symptom severity is multi-factorial, with phenotypic expression varying amongst individuals within the same family. Genetic testing is another measure of identifying the underlying mutations which cause the pathologic symptoms of the disease. This mode of testing is available at select laboratories.[10]
Work-up to exclude the differential diagnosis of the electrolyte abnormalities is key.[14]
- In Gitelman syndrome hypocalciuria is present, and a urine calcium:creatinine ratio may help distinguish it from Bartter syndrome as the two disorders can be clinically indistinguishable. Additionally in Bartter syndrome maximal urine concentrating ability is lost.
- Laxative abuse can mimic the serum electrolyte abnormalities, but fractional excretion of potassium will be low
- Diuretic abuse could be suspected if urinary chloride excretion varies by time of day but may require a diuretic assay to detect
- Surreptitious vomiting can cause metabolic alkalosis and hypokalaemia, but urinary chloride levels will be low
- Medication history; Proton-pump inhibitors can cause an isolated hypomagnesaemia phenotype, and aminoglycosides such as gentamicin can cause a transient metabolic alkalosis with hypokalaemia and hypomagnesaemia that resolves 2–6 weeks after drug termination.
- Primary aldosteronism will cause metabolic alkalosis and hypokalaemia, but hypertension will be present and serum renin will be low
- EAST syndrome, though neurological features will predominate
- Renal cysts and diabetes syndrome can cause hypomagnesaemia and hypocalcuria, but is distinguished by early onset chronic kidney disease and an autosomal dominant inheritance pattern of renal cysts and/or diabetes
Treatment
Most asymptomatic individuals with Gitelman syndrome can be monitored without medical treatment.[2] Dietary modification of a high salt diet incorporated with,[10] potassium and magnesium supplementation to normalize blood levels is the mainstay of treatment.[2] Large doses of potassium and magnesium are often necessary to adequately replace the electrolytes lost in the urine.[2] Diarrhea is a common side effect of oral magnesium which can make replacement by mouth difficult but dividing the dose to 3-4 times a day is better tolerated.[2] Severe deficits of potassium and magnesium require intravenous replacement. If low blood potassium levels are not sufficiently replaced with replacement by mouth, aldosterone antagonists (such as spironolactone or eplerenone) or epithelial sodium channel blockers such as amiloride can be used to decrease urinary wasting of potassium.[2]
In patients with early onset of the disease such as infants and children, indomethacin is the drug of choice utilized to treat growth disturbances.[10] Indomethacin in a study by Blanchard et al. 2015 was shown to increase serum potassium levels, and decrease renin concentration. Adverse effects of indomethacin include a decrease in the glomerular filtration rate, and gastrointestinal disturbances.[15]
Cardiac evaluation is promoted in the prevention of dysrhythmias and monitoring of QT interval activity.[10] Medications that extend or prolong the QT interval (macrolides, antihistamines, beta-2 agonists) should be avoided in these patients to prevent cardiac death.[3]
Epidemiology
Gitelman syndrome is estimated to have a prevalence of 1 in 40,000 homozygous people .[2] The ratio of men to women affected is 1:1. This disease is encountered typically past the 1st decade of life, during adolescence or adulthood but can occur in the neonatal period. Heterozygous carriers of the SLC12A3 gene mutations are 1% of the population.[10] Parents with Gitelman syndrome have a low probability of passing the disorder to their offspring roughly 1 in 400 unless they are both carriers of the disease.[9]
History
The condition is named for Hillel Jonathan Gitelman (1932– January 12, 2015), an American nephrologist working at University of North Carolina School of Medicine. He first described the condition in 1966, after observing a pair of sisters with the disorder. Gitelman and his colleagues later identified and isolated the gene responsible (SLC12A3) by molecular cloning.[16][17][18][19][20]
References
- Fischer, Artwork by Holly (2013-01-31), English: This is an image of a kidney nephron and its structure., retrieved 2020-04-01
- Nakhoul, F; Nakhoul, N; Dorman, E; Berger, L; Skorecki, K; Magen, D (February 2012). "Gitelman's syndrome: a pathophysiological and clinical update". Endocrine (Review). 41 (1): 53–7. doi:10.1007/s12020-011-9556-0. PMID 22169961. S2CID 5820317.
- Seyberth, Hannsjörg W.; Schlingmann, Karl P. (October 2011). "Bartter- and Gitelman-like syndromes: salt-losing tubulopathies with loop or DCT defects". Pediatric Nephrology. 26 (10): 1789–1802. doi:10.1007/s00467-011-1871-4. ISSN 0931-041X. PMC 3163795. PMID 21503667.
- Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP (June 1996). "Bartter's syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2". Nat. Genet. 13 (2): 183–8. doi:10.1038/ng0696-183. PMID 8640224. S2CID 42296304.
- O'Shaughnessy KM, Karet FE (2004). "Salt handling and hypertension". J. Clin. Invest. 113 (8): 1075–81. doi:10.1172/JCI21560. PMC 385413. PMID 15085183.
- Pieter Du Toit van der Merwe, Megan A. Rensburg, William L. Haylett, Soraya Bardien, and M. Razeen Davids (2017). "Gitelman syndrome in a South African family presenting with hypokalaemia and unusual food cravings". BMC Nephrol. 18 (38): 38. doi:10.1186/s12882-017-0455-3. PMC 5270235. PMID 28125972.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Cruz, Dinna N.; Shaer, Andrea J.; Bia, Margaret J.; Lifton, Richard P.; Simon, David B. (February 2001). "Gitelman's syndrome revisited: An evaluation of symptoms and health-related quality of life". Kidney International. 59 (2): 710–717. doi:10.1046/j.1523-1755.2001.059002710.x. ISSN 0085-2538. PMID 11168953.
- Riveira-Munoz, Eva; Chang, Qing; Godefroid, Nathalie; Hoenderop, Joost G.; Bindels, René J.; Dahan, Karin; Devuyst, Olivier; Belgian Network for Study of Gitelman Syndrome (April 2007). "Transcriptional and functional analyses of SLC12A3 mutations: new clues for the pathogenesis of Gitelman syndrome". Journal of the American Society of Nephrology. 18 (4): 1271–1283. doi:10.1681/ASN.2006101095. ISSN 1046-6673. PMID 17329572.
- Knoers, Nine VAM Levtchenko, Elena N (2008-07-30). "Gitelman syndrome". Orphanet Journal of Rare Diseases. BioMed Central Ltd. 3: 22. doi:10.1186/1750-1172-3-22. OCLC 804470918. PMC 2518128. PMID 18667063.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - "Gitelman Syndrome". NORD (National Organization for Rare Disorders). Retrieved 2020-03-29.
- "Gitelman Syndrome". The Lecturio Medical Concept Library. Retrieved 23 July 2021.
- Viering D, Schlingmann KP, Hureaux M, Nijenhuis T, Mallett A, Chan M, van Beek A, van Eerde A, Coulibaly JM, Vallet M, Decramer S, Pelletier S, Klaus G, Kömhoff M, Beetz R, Patel C, Shenoy M, Steenbergen E, Anderson G, Bongers E, Bergmann C, Panneman D, Rodenburg R, Kleta R, Houillier P, Konrad M, Vargas-Poussou R, Knoers N, Bockenhauer D, de Baaij J (2021). "Gitelman-Like Syndrome Caused by Pathogenic Variants in mtDNA". J. Am. Soc. Nephrol. 33 (2): 305–325. doi:10.1681/ASN.2021050596. PMC 8819995. PMID 34607911.
- Viganò, Cristina; Amoruso, Chiara; Barretta, Francesco; Minnici, Giuseppe; Albisetti, Walter; Syrèn, Marie-Louise; Bianchetti, Mario G.; Bettinelli, Alberto (2013-01-01). "Renal phosphate handling in Gitelman syndrome—the results of a case–control study" (PDF). Pediatric Nephrology. 28 (1): 65–70. doi:10.1007/s00467-012-2297-3. ISSN 1432-198X. PMID 22990302. S2CID 13727845.
- Urwin, Stephanie; Willows, Jamie; Sayer, John A. (2020). "The challenges of diagnosis and management of Gitelman syndrome". Clinical Endocrinology. 92 (1): 3–10. doi:10.1111/cen.14104. ISSN 1365-2265. PMID 31578736.
- Blanchard, Anne; Vargas-Poussou, Rosa; Vallet, Marion; Caumont-Prim, Aurore; Allard, Julien; Desport, Estelle; Dubourg, Laurence; Monge, Matthieu; Bergerot, Damien; Baron, Stéphanie; Essig, Marie (February 2015). "Indomethacin, Amiloride, or Eplerenone for Treating Hypokalemia in Gitelman Syndrome". Journal of the American Society of Nephrology. 26 (2): 468–475. doi:10.1681/ASN.2014030293. ISSN 1046-6673. PMC 4310664. PMID 25012174.
- synd/2329 at Who Named It?
- Gitelman HJ, Graham JB, Welt LG (1966). "A new familial disorder characterized by hypokalemia and hypomagnesemia". Trans. Assoc. Am. Physicians. 79: 221–35. PMID 5929460.
- Unwin RJ, Capasso G (2006). "Bartter's and Gitelman's syndromes: their relationship to the actions of loop and thiazide diuretics" (PDF). Current Opinion in Pharmacology. 6 (2): 208–213. doi:10.1016/j.coph.2006.01.002. PMID 16490401. Archived from the original (PDF) on 2013-10-23.
- "Dr. Hillel Jonathan Gitelman". The News & Observer. Retrieved 5 March 2018.
- "Hillel J. Gitelman '54". Princeton Alumni Weekly. May 13, 2015. Retrieved 5 March 2018.
{kind=link}
External links
- "Gitelman syndrome". MedlinePlus. U.S. National Library of Medicine.