Hereditary motor and sensory neuropathy
Hereditary motor and sensory neuropathies (HMSN) is a name sometimes given to a group of different neuropathies which are all characterized by their impact upon both afferent and efferent neural communication. HMSN are characterised by atypical neural development and degradation of neural tissue. The two common forms of HMSN are either hypertrophic demyelinated nerves or complete atrophy of neural tissue. Hypertrophic condition causes neural stiffness and a demyelination of nerves in the peripheral nervous system, and atrophy causes the breakdown of axons and neural cell bodies.[1] In these disorders, a patient experiences progressive muscle atrophy and sensory neuropathy of the extremities.[2]
| Hereditary motor and sensory neuropathy | |
|---|---|
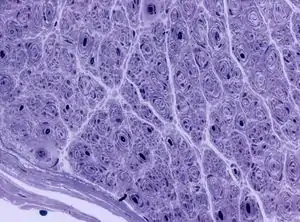 | |
| Onion bulb formations in a nerve biopsy in a case of HMSN type I | |
| Specialty | Neurology |
The term "hereditary motor and sensory neuropathy" was used mostly historically to denote the more common forms Charcot–Marie–Tooth disease (CMT). With the identification of a wide number of genetically and phenotypically distinct forms of CMT, the term HMSN is now used less frequently.
Symptoms and signs

Neuropathy disorders usually have onset in childhood or young adulthood. Motor symptoms seem to be more predominant than sensory symptoms.[2] Symptoms of these disorders include: fatigue, pain, lack of balance, lack of feeling, lack of reflexes, and lack of sight and hearing, which result from muscle atrophy. Patients can also have high arched feet, hammer toes, foot drop, foot deformities, and scoliosis. These symptoms are a result of severe muscular weakness and atrophy. In patients with demyelinating neuropathy, symptoms are due to slow nerve conduction velocities; however people with axonal degradation have average-to-normal nerve conduction velocities.
Causes
All hereditary motor and sensory neuropathies are inherited. Chromosomes 17 and 1 seem to be the most common chromosomes with mutations.[1] The disease can be inherited in an autosomal dominant, autosomal recessive or X-linked manner.
Diagnosis
Patients with hereditary motor and sensory neuropathies are diagnosed through a physical evaluation that looks for muscle atrophy, weakness, and sensory responses.[3] In addition to this, electromyography and motor nerve conduction tests can help clinicians decide what type of motor and sensory neuropathy it is and how severe the disease is. Final confirmation can come through genetic testing.
Classification
Charcot–Marie–Tooth disease was first described in 1886 by Jean-Martin Charcot, Pierre Marie, and independently Howard Henry Tooth.[2] In the 1950s, further classification occurred and separated patients into two distinct groups. Group one was characterized by slow nerve conduction velocities and demyelinating neuropathy. Group two was characterized by mostly normal nerve conduction velocities and degeneration of axons. In 1968, HMSN were classified again into seven groups:[1][4]
| Type | Other names | Diseases | OMIM | |
|---|---|---|---|---|
| HMSN1 | Charcot–Marie–Tooth disease type 1A and 1B | 5815 | (multiple) | Hypertrophic demyelinating type: affected individuals experience weakness and atrophy in the lower legs in adolescence, and later develop weakness in the hands. This is the most common type of CMT. |
| HMSN2 | Charcot–Marie–Tooth disease type 2 | 2343 | (multiple) | Neuronal type: symptoms similar to type1, onset in adolescence. |
| HMSN3 | Dejerine–Sottas disease (Charcot–Marie–Tooth type 3) | 5821 | 145900 | Onset in infancy and results in delayed motor skills, much more severe than types 1 & 2. |
| HMSN4 | Refsum disease | 11213 | 266500 | Spinal type: Muscle weakness and atrophy as in other types of CMT, but set apart by being autosomal recessive inheritance. |
| HMSN5 | Charcot–Marie–Tooth with pyramidal features | — | 600361 | Pyramidal type: onset between ages 5–12. Lower legs are affected first by muscle weakness and atrophy followed by the upper extremities. Type 5 is also associated with visual and hearing loss. |
| HMSN6 | Charcot–Marie–Tooth type 6 | 32095 | 601152 | Early onset muscular weakness and atrophy followed by optic atrophy resulting in vision loss and possibly blindness. |
| HMSN7 | HMSN+retinitis pigmentosa | 32094 | — | Later onset with muscular weakness and atrophy mostly in the lower extremities. |
Treatment
There is currently no known pharmacological treatment to hereditary motor and sensory neuropathy. However, the majority of people with these diseases are able to walk and be self-sufficient.[3] Some methods of relief for the disease include physical therapy, stretching, braces, and sometimes orthopedic surgery. Since foot disorders are common with neuropathy, precautions must be taken to strengthen these muscles and use preventative care and physical therapy to prevent injury and deformities.
Prognosis
Hereditary motor and sensory neuropathy are relatively common and are often inherited with other neuromuscular conditions, and these comorbidities cause an accelerated progression of the disease.
Most forms of HMSN affect males earlier and more severely than females, but others show no predilection to either sex. HMSN affects all ethnic groups, with the most common forms having no racial predilection, but other recessively inherited forms tending to impact specific ethnic groups. Onset of HMSN is most common in early childhood, with clinical effects occurring before the age of 10, but some symptoms are lifelong and progress slowly. Therefore, these symptoms do not appear until later in life.[1]
See also
References
- Charcot-Marie-Tooth and Other Hereditary Motor and Sensory Neuropathies at eMedicine
- Vance, Jeffery M. (1991). "Hereditary motor and sensory neuropathies". Journal of Medical Genetics. 28 (1): 1–5. doi:10.1136/jmg.28.1.1. PMC 1016739. PMID 1999826.
- American Association of Neuromuscular & Electrodiagnostic Medicine. (2013). Hereditary Motor Sensory Neuropathy. http://www.aanem.org/Education/Patient-Resources/Disorders/Hereditary-Motor-Sensory-Neuropathy.aspx Accessed on 11/10/13.
- Dyck, Peter James; Lambert, Edward H. (1968). "Lower Motor and Primary Sensory Neuron Diseases With Peroneal Muscular Atrophy". Archives of Neurology. 18 (6): 603–18. doi:10.1001/archneur.1968.00470360025002. PMID 4297451.
Further reading
- Reilly MM (October 2000). "Classification of the hereditary motor and sensory neuropathies". Curr. Opin. Neurol. 13 (5): 561–4. doi:10.1097/00019052-200010000-00009. PMID 11073363. S2CID 43241647.