Dejerine–Sottas disease
Dejerine–Sottas disease, also known as, Dejerine–Sottas neuropathy, progressive hypertrophic interstitial polyneuropathy of childhood and onion bulb neuropathy[1] (and, hereditary motor and sensory polyneuropathy type III and Charcot–Marie–Tooth disease type 3), is a hereditary neurological disorder characterised by damage to the peripheral nerves and resulting progressive muscle wasting. The condition is caused by mutations in a various genes and currently has no known cure.[2]
| Dejerine–Sottas syndrome | |
|---|---|
| Other names | Charcot–Marie–Tooth disease type 3 |
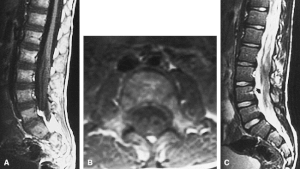 | |
| MRI compatible with Dejerine-Sottas type spinal nerve enlargement | |
| Specialty | Neurology |
| Usual onset | Infancy or early childhood |
The disorder is named for Joseph Jules Dejerine and Jules Sottas, French neurologists who first described it.[3][4]
Signs and symptoms
Onset occurs in infancy or early childhood, usually before three years of age. Progression is slow until the teenage years at which point it may accelerate, resulting in severe disability.
Symptoms are usually more severe and rapidly progressive than in the other more common Charcot–Marie–Tooth diseases. Some patients may never walk and solely use wheelchairs by the end of their first decade, while others may need only a cane (walking stick) or similar support through life.
Dejerine–Sottas disease is characterized by moderate to severe lower and upper extremity weakness and loss of sensation, which occur mainly in the lower legs, forearms, feet and hands. Loss of muscle mass and reduced muscle tone can occur as the disease progresses. Other symptoms may include pain in the extremities, curvature of the spine, clawed hands, foot deformities, ataxia, peripheral areflexia, and slow acquisition of motor skills in childhood. Symptoms that are less common can include limitation of eye movements, other eye problems such as nystagmus or anisocoria, or mild hearing loss.[2]
Causes
Dejerine–Sottas neuropathy is caused by a genetic defect either in the proteins found in axons or the proteins found in myelin.[2] Specifically, it has been associated with mutations in MPZ,[5] PMP22,[6] PRX,[7] and EGR2[8] genes. The disorder is inherited in an autosomal dominant or autosomal recessive manner.[2]
Diagnosis
On medical imaging, the nerves of the extremities (and cranial nerves in some cases) appear enlarged due to hypertrophy of the connective interstitial tissue, giving the nerves a distinct "onion-bulb" appearance. Peripheral (and possibly cranial) nerve excitability and conduction speed are reduced.
Treatment
Management is symptomatic for this condition .
See also
References
- Satran, R. (1980). "Dejerine-Sottas Disease Revisited". Archives of Neurology. 37 (2): 67–68. doi:10.1001/archneur.1980.00500510025002. PMID 7356409.
- Muscular Dystrophy Association (2012). "Dejerine-Sottas Disease". Retrieved 7 May 2012.
- synd/1736 at Who Named It?
- Dejerine, J. J.; Sottas, J. (1893). "Sur la névrite interstitielle hypertrophique et progressive de l'enfance; affection souvent familiale et à debut infantile, caractérisée par une atrophie musculaire des extrémities, avec troubles marqués de la sensibilité et ataxie des mouvements et relevant d'une névrite interstitielle hypertrophique a marche ascendante avec lésions médullaires consécutives" (PDF). Comptes Rendus des Séances de la Société de Biologie. Paris. 45: 63–96.
- Hayasaka K, Himoro M, Sawaishi Y, et al. (November 1993). "De novo mutation of the myelin P0 gene in Dejerine-Sottas disease (hereditary motor and sensory neuropathy type III)". Nat. Genet. 5 (3): 266–8. doi:10.1038/ng1193-266. PMID 7506095. S2CID 2512684.
- Roa BB, Dyck PJ, Marks HG, Chance PF, Lupski JR (November 1993). "Dejerine-Sottas syndrome associated with point mutation in the peripheral myelin protein 22 (PMP22) gene". Nat. Genet. 5 (3): 269–73. doi:10.1038/ng1193-269. PMID 8275092. S2CID 6407166.
- Kabzinska D, Drac H, Sherman DL, et al. (March 2006). "Charcot-Marie-Tooth type 4F disease caused by S399fsx410 mutation in the PRX gene". Neurology. 66 (5): 745–7. doi:10.1212/01.wnl.0000201269.46071.35. PMID 16534116. S2CID 22585765.
- Boerkoel CF, Takashima H, Bacino CA, Daentl D, Lupski JR (July 2001). "EGR2 mutation R359W causes a spectrum of Dejerine-Sottas neuropathy". Neurogenetics. 3 (3): 153–7. doi:10.1007/s100480100107. PMID 11523566. S2CID 32746701.