Palmoplantar keratoderma
Palmoplantar keratodermas are a heterogeneous group of disorders characterized by abnormal thickening of the stratum corneum of the palms and soles.
| Palmoplantar keratoderma | |
|---|---|
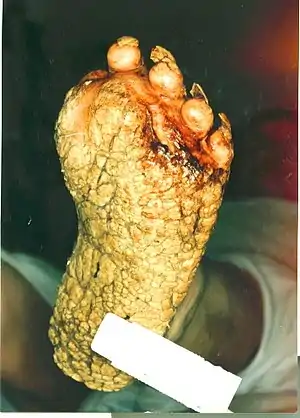 | |
| Patient with severe plantar keratosis. | |
| Specialty | Dermatology |
Autosomal recessive, dominant, X-linked, and acquired forms have all been described.[1]: 505 [2]: 211 [3]
Types
Clinically, three distinct patterns of palmoplantar keratoderma may be identified: diffuse, focal, and punctate.[1]: 505
Diffuse
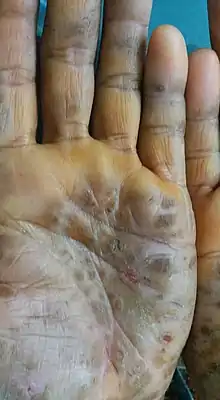
Diffuse palmoplantar keratoderma is a type of palmoplantar keratoderma that is characterized by an even, thick, symmetric hyperkeratosis over the whole of the palm and sole, usually evident at birth or in the first few months of life.[1]: 505 Restated, diffuse palmoplantar keratoderma is an autosomal dominant disorder in which hyperkeratosis is confined to the palms and soles.[4] The two major types can have a similar clinical appearance:[4]
- Diffuse epidermolytic palmoplantar keratoderma (also known as "Palmoplantar keratoderma cum degeneratione granulosa Vörner," "Vörner's epidermolytic palmoplantar keratoderma", and "Vörner keratoderma"[4]) is one of the most common patterns of palmoplantar keratoderma, an autosomal dominant condition that presents within the first few months of life, characterized by a well-demarcated, symmetric thickening of palms and soles, often with a "dirty" snakeskin appearance due to underlying epidermolysis.[1]: 506
- Diffuse nonepidermolytic palmoplantar keratoderma (also known as "Diffuse orthohyperkeratotic keratoderma," "Hereditary palmoplantar keratoderma," "Keratosis extremitatum progrediens," "Keratosis palmoplantaris diffusa circumscripta," "Tylosis," "Unna–Thost disease", and "Unna–Thost keratoderma"[4]) is inherited as an autosomal dominant condition and is present from infancy, characterized by a well-demarcated, symmetric, often "waxy" keratoderma involving the whole of the palms and soles.[1]: 506–8 [2]: 213
Focal
Focal palmoplantar keratoderma, a type of palmoplantar keratoderma in which large, compact masses of keratin develop at sites of recurrent friction, principally on the feet, although also on the palms and other sites, a pattern of calluses that may be discoid (nummular) or linear.
- Focal palmoplantar keratoderma with oral mucosal hyperkeratosis (also known as "Focal epidermolytic palmoplantar keratoderma,"[4] "Hereditary painful callosities,"[4][5] "Hereditary painful callosity syndrome,"[1] "Keratosis follicularis,"[1] "Keratosis palmoplantaris nummularis",[1] and "Nummular epidermolytic palmoplantar keratoderma"[4]) is an autosomal dominant keratoderma that represents a clinical overlap syndrome with pachyonychia congenita type I but without the classic nail involvement.[1]: 510
Punctate
Punctate palmoplantar keratoderma is a form of palmoplantar keratoderma in which many tiny "raindrop" keratoses involve the palmoplantar surface, skin lesions which may involve the whole of the palmoplantar surface, or may be more restricted in their distribution.[1]: 505 [4]
- Type 1: Keratosis punctata palmaris et plantaris (also known as "Autosomal-dominant hereditary punctate keratoderma associated with malignancy," "Buschke–Fischer–Brauer disease," "Davis Colley disease," "Keratoderma disseminatum palmaris et plantaris," "Keratosis papulosa," "Keratoderma punctatum," "Keratodermia punctata," "Keratoma hereditarium dissipatum palmare et plantare," "Palmar and plantar seed dermatoses," "Palmar keratoses," "Papulotranslucent acrokeratoderma," "Punctate keratoderma," "Punctate keratoses of the palms and soles," and "Maculosa disseminata") is a skin condition, an autosomal dominant palmoplantar keratoderma with variable penetrance, characterized clinically by multiple, tiny, punctate keratoses over the entire palmoplantar surfaces, beginning over the lateral edge of the digits.[1]: 509 [2]: 212–213 It has been linked to 15q22-q24.[6]
- Type 2: Spiny keratoderma (also known as "Porokeratosis punctata palmaris et plantaris," "Punctate keratoderma," and "Punctate porokeratosis of the palms and soles") is an autosomal dominant keratoderma of late onset that develops in patients aged 12 to 50, characterized by multiple tiny keratotic plugs, resembling the spines on a music box, involving the entire palmoplantar surfaces.[1]: 509 [4]
- Type 3: Focal acral hyperkeratosis (also known as "Acrokeratoelastoidosis lichenoides," and "Degenerative collagenous plaques of the hand") is a late-onset keratoderma, inherited as an autosomal dominant condition, characterized by oval or polygonal crateriform papules developing along the border of the hands, feet, and wrists.[1]: 509 It is considered similar to Costa acrokeratoelastoidosis.[7]
Ungrouped
- Palmoplantar keratoderma and spastic paraplegia (also known as "Charcot–Marie–Tooth disease with palmoplantar keratoderma and nail dystrophy"[1]) is an autosomal dominant or x-linked dominant condition that begins in early childhood with thick focal keratoderma over the soles and, to a lesser extent, the palms.[1]: 513
- Palmoplantar keratoderma of Sybert (also known as "Greither palmoplantar keratoderma,"[1] "Greither syndrome,"[4] "Keratosis extremitatum hereditaria progrediens,"[1] "Keratosis palmoplantaris transgrediens et progrediens"[1] "Sybert keratoderma,"[4] and "Transgrediens and progrediens palmoplantar keratoderma"[4]) is an extremely rare autosomal dominant[8] keratoderma (a skin condition involving horn-like growths) with symmetric severe involvement of the whole palmoplantar surface in a glove-and-stocking distribution.[1]: 509 It was characterized by Aloys Greither in 1952.[9][10][11] It was characterized by Virginia Sybert in 1988.[12] An autosomal recessive form which is known as Mal de Meleda has been described.[13] This is associated with mutations in the Secreted Ly-6/uPAR-related protein 1 (SLURP1) gene.
- Striate palmoplantar keratoderma (also known as "Acral keratoderma,"[1] "Brünauer-Fuhs-Siemens type of palmoplantar keratoderma,"[1] "Focal non-epidermolytic palmoplantar keratoderma,"[4] "Keratosis palmoplantaris varians,"[1] "Palmoplantar keratoderma areata,"[4] "Palmoplantar keratoderma striata,"[4] "Wachter keratoderma,"[4]: 778, 785 and "Wachters palmoplantar keratoderma"[1]) is a cutaneous condition, an autosomal dominant keratoderma principally involving the soles with onset in infancy or the first few years of life.[1]: 509
- Carvajal syndrome (also known as "Striate palmoplantar keratoderma with woolly hair and cardiomyopathy"[4] and "Striate palmoplantar keratoderma with woolly hair and left ventricular dilated cardiomyopathy,"[1]) is a cutaneous condition inherited in an autosomal recessive fashion, and due to a defect in desmoplakin.[4]: 811 Striate palmoplantar keratoderma, woolly hair, and left ventricular dilated cardiomyopathy has been described in both autosomal dominant and autosomal recessive forms, but only the recessive forms have a clear association with dilated cardiomyopathy.[1]: 513 The skin disease presents as a striate palmoplantar keratoderma with some nonvolar involvement, particularly at sites of pressure or abrasion.[1]: 513
- Scleroatrophic syndrome of Huriez (also known as "Huriez syndrome," "Palmoplantar keratoderma with scleroatrophy,"[4] "Palmoplantar keratoderma with sclerodactyly," "Scleroatrophic and keratotic dermatosis of the limbs," and "Sclerotylosis") is an autosomal dominant keratoderma with sclerodactyly present at birth with a diffuse symmetric keratoderma of the palms and soles.[1]: 513 [2]: 576 An association with 4q23 has been described.[14] It was characterized in 1968.[15]
- Vohwinkel syndrome (also known as "Keratoderma hereditaria mutilans,"[4] "Keratoma hereditaria mutilans,"[4] "Mutilating keratoderma of Vohwinkel",[2]: 213 "Mutilating palmoplantar keratoderma"[4]) is a diffuse autosomal dominant keratoderma with onset in early infancy characterized by a honeycombed keratoderma involving the palmoplantar surfaces.[1]: 512 Mild to moderate sensorineural hearing loss is often associated.[1] It has been associated with GJB2.[16] It was characterized in 1929.[17]
- Olmsted syndrome (also known as "Mutilating palmoplantar keratoderma with periorificial keratotic plaques," "Mutilating palmoplantar keratoderma with periorificial plaques"[4] and "Polykeratosis of Touraine") is a keratoderma of the palms and soles, with flexion deformity of the digits, that begins in infancy.[1]: 510 [2]: 214 [4][18] Treatment with retinoids has been described.[19] It has been associated with mutations in TRPV3.[20]
- Aquagenic keratoderma, also known as acquired aquagenic palmoplantar keratoderma,[4]: 788 transient reactive papulotranslucent acrokeratoderma,[4] aquagenic syringeal acrokeratoderma,[4] and aquagenic wrinkling of the palms,[2] is a skin condition characterized by the development of white papules on the palms after water exposure.[2]: 215 The condition causes irritation of the palms when touching certain materials after being wet, e.g., paper, cloth. An association with cystic fibrosis has been suggested.[21] The association with cystic fibrosis suggests an increased salt content in the skin.[22]
Genetics
Epidermolytic palmoplantar keratoderma has been associated with keratin 9 and keratin 16.[23]
Nonepidermolytic palmoplantar keratoderma has been associated with keratin 1 and keratin 16.[24]
Treatment
Keratolytic products containing, urea, salicylic, glycolic and lactic acids are helpful. One proprietary cream (Pedifix Cracks and Calluses Cream) is efficacious.
See also
References
- Freedberg IM, Fitzpatrick TB (2003). Fitzpatrick's Dermatology in General Medicine (6th ed.). New York; London: McGraw-Hill. ISBN 978-0-07-138076-8.
- James W, Berger T, Elston D (2005). Andrews' Diseases of the Skin: Clinical Dermatology (10th ed.). Saunders. ISBN 978-0-7216-2921-6.
- Patel S, Zirwas M, English JC (2007). "Acquired palmoplantar keratoderma". American Journal of Clinical Dermatology. 8 (1): 1–11. doi:10.2165/00128071-200708010-00001. PMID 17298101.
- Rapini RP, Bolognia JL, Jorizzo JL (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. p. 740. ISBN 978-1-4160-2999-1.
- Ryan P, Baird G, Benfanti P (March 2007). "Hereditary painful callosities: case report and review of the literature". Foot & Ankle International. 28 (3): 377–8. doi:10.3113/FAI.2007.0377. PMID 17371662.
- Martinez-Mir A, Zlotogorski A, Londono D, Gordon D, Grunn A, Uribe E, Horev L, Ruiz IM, Davalos NO, Alayan O, Liu J, Gilliam TC, Salas-Alanis JC, Christiano AM (December 2003). "Identification of a locus for type I punctate palmoplantar keratoderma on chromosome 15q22-q24". Journal of Medical Genetics. 40 (12): 872–8. doi:10.1136/jmg.40.12.872. PMC 1735333. PMID 14684683.
- Erkek E, Koçak M, Bozdoğan O, Atasoy P, Birol A (2004). "Focal acral hyperkeratosis: a rare cutaneous disorder within the spectrum of Costa acrokeratoelastoidosis". Pediatric Dermatology. 21 (2): 128–30. doi:10.1111/j.0736-8046.2004.21208.x. PMID 15078352.
- Leonard AL, Freedberg IM (October 2003). "Palmoplantar keratoderma of Sybert". Dermatology Online Journal. 9 (4): 30. PMID 14594603.
- synd/1800 at Who Named It?
- Greither A (May 1952). "[Keratosis extremitatum hereditaria progrediens with genetic dominant]". Der Hautarzt; Zeitschrift für Dermatologie, Venerologie, und Verwandte Gebiete. 3 (5): 198–203. PMID 14945735.
- Gach JE, Munro CS, Lane EB, Wilson NJ, Moss C (November 2005). "Two families with Greither's syndrome caused by a keratin 1 mutation". Journal of the American Academy of Dermatology. 53 (5 Suppl 1): S225-30. doi:10.1016/j.jaad.2005.01.139. PMID 16227096.
- Sybert VP, Dale BA, Holbrook KA (January 1988). "Palmar-plantar keratoderma. A clinical, ultrastructural, and biochemical study". Journal of the American Academy of Dermatology. 18 (1 Pt 1): 75–86. doi:10.1016/S0190-9622(88)70012-2. PMID 2450111.
- Gurel G, Cilingir O, Kutluay O, Arslan S, Sahin S, Colgecen E (2019) Patient with Mal de Meleda in whom a novel gene mutation was identified. Eurasian J Med 51(2):206–208
- Lee YA, Stevens HP, Delaporte E, Wahn U, Reis A (January 2000). "A gene for an autosomal dominant scleroatrophic syndrome predisposing to skin cancer (Huriez syndrome) maps to chromosome 4q23". American Journal of Human Genetics. 66 (1): 326–30. doi:10.1086/302718. PMC 1288338. PMID 10631162.
- Huriez C, Deminatti M, Agache P, Mennecier M (February 1968). "[A gene dysplasia not previously known: frequently degenerative sclero-atrophying and keratodermic genodermatosis of the extremities]". La Semaine des Hopitaux (in French). 44 (8): 481–8. PMID 4298032.
- Maestrini E, Korge BP, Ocaña-Sierra J, Calzolari E, Cambiaghi S, Scudder PM, Hovnanian A, Monaco AP, Munro CS (July 1999). "A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel's syndrome) in three unrelated families". Human Molecular Genetics. 8 (7): 1237–43. doi:10.1093/hmg/8.7.1237. PMID 10369869.
- Vohwinkel KH (1929). "Keratoma hereditarium mutilans". Archiv für Dermatologie und Syphilis. 158 (2): 354–364. doi:10.1007/bf01826619.
- Kumar P, Sharma PK, Kar HK (2008). "Olmsted syndrome". Indian Journal of Dermatology. 53 (2): 93–5. doi:10.4103/0019-5154.41657. PMC 2763718. PMID 19881998.
- Dessureault J, Poulin Y, Bourcier M, Gagne E (2003). "Olmsted syndrome-palmoplantar and periorificial keratodermas: association with malignant melanoma". Journal of Cutaneous Medicine and Surgery. 7 (3): 236–42. doi:10.1007/s10227-002-0107-4. PMID 12704531.
- Lin Z, Chen Q, Lee M, Cao X, Zhang J, Ma D, Chen L, Hu X, Wang H, Wang X, Zhang P, Liu X, Guan L, Tang Y, Yang H, Tu P, Bu D, Zhu X, Wang K, Li R, Yang Y (March 2012). "Exome sequencing reveals mutations in TRPV3 as a cause of Olmsted syndrome". American Journal of Human Genetics. 90 (3): 558–64. doi:10.1016/j.ajhg.2012.02.006. PMC 3309189. PMID 22405088.
- Garçon-Michel N, Roguedas-Contios AM, Rault G, Le Bihan J, Ramel S, Revert K, Dirou A, Misery L (July 2010). "Frequency of aquagenic palmoplantar keratoderma in cystic fibrosis: a new sign of cystic fibrosis?". The British Journal of Dermatology. 163 (1): 162–6. doi:10.1111/j.1365-2133.2010.09764.x. PMID 20302572.
- Sezer E, Durmaz EÖ, Çetin E, Şahin S (2015). "Permanent treatment of aquagenic syringeal acrokeratoderma with endoscopic thoracic sympathectomy". Indian Journal of Dermatology, Venereology and Leprology. 81 (6): 648–50. doi:10.4103/0378-6323.168331. PMID 26515860.
- Online Mendelian Inheritance in Man (OMIM): 144200
- Online Mendelian Inheritance in Man (OMIM): 600962