Dyskeratosis congenita
Dyskeratosis congenita (DKC), also known as Zinsser-Engman-Cole syndrome, is a rare progressive congenital disorder with a highly variable phenotype.[3] The entity was classically defined by the triad of abnormal skin pigmentation, nail dystrophy, and leukoplakia of the oral mucosa, but these components do not always occur.[3] DKC is characterized by short telomeres. Some of the manifestations resemble premature ageing (similar to progeria). The disease initially mainly affects the skin, but a major consequence is progressive bone marrow failure which occurs in over 80%, causing early mortality.[3]
| Dyskeratosis congenita | |
|---|---|
| Other names | Zinsser-Cole-Engman syndrome,[1][2]: 570 |
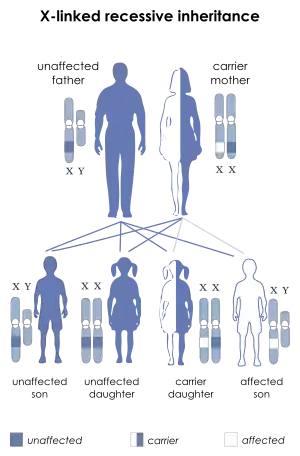 | |
| Dyskeratosis congenital is inherited in an X-linked recessive manner | |
| Specialty | Medical genetics |
Presentation
DKC can be characterized by cutaneous pigmentation, premature graying, dystrophy of the nails, leukoplakia of the oral mucosa, continuous lacrimation due to atresia of the lacrimal ducts, often thrombocytopenia, anemia, testicular atrophy in the male carriers, and predisposition to cancer.[4] Many of these symptoms are characteristic of geriatrics, and those carrying the more serious forms of the disease often have significantly shortened lifespans. Also, liver abnormalities are associated with this syndrome, Nodular Regenerative Hypoplasia of the liver, although rare, it is one of many manifestations of liver disorders short telomeres can cause.[5]
Predisposition to cancer
Susceptibility to cancer seems counterintuitive because in many known cancers reactivation of telomerase is actually a required step for malignancy to evolve (see telomere). In a disease where telomerase is affected, it does not seem to follow that cancer would be a complication to result. The authors note the paradoxical nature of cancer predisposition in individuals who seem to lack one of the required components for cancer to form. It is thought [6] that without functional telomerase, chromosomes will likely be attached together at their ends through the non-homologous end joining pathway. If this proves to be a common enough occurrence, malignancy even without telomerase present is possible. Myelodysplastic Syndrome is associated with this syndrome usually presenting as a Hypoplastic Bone Marrow that can resemble Aplastic Anemia, but can be differentiated with >10% dysplasia in affected cell lines, sometimes not possible though because of the Hypoplastic marrow reducing blood cells to be observed, genetic clones are usually not present more often than not with Hypoplastic Myelodysplastic Disorder associated with Dyskeratosis Congenita.
Genetics
Of the components of the telomerase RNA component (TERC), one of key importance is the box H/ACA domain. This H/ACA domain is responsible for maturation and stability of TERC and therefore of telomerase as a whole. The mammalian H/ACA ribonucleoprotein contains four protein subunits: dyskerin, Gar1, Nop10, and Nhp2. Mutations in Nop10,[7] Nhp2[8] and dyskerin1[9] have all been shown to lead to DKC-like symptoms.
X-linked
The best characterized form of dyskeratosis congenita is a result of one or more mutations in the long arm of the X chromosome in the gene DKC1.[6][9] This results in the X-linked recessive form of the disease wherein the major protein affected is dyskerin. Of the five mutations described by Heiss and colleagues in Nature Genetics,[9] four were single nucleotide polymorphisms all resulting in the change of highly conserved amino acids. One case was an in-frame deletion resulting in the loss of a leucine residue, also conserved in mammals. In three of the cases, the specific amino acids affected (phenylalanine, proline, glycine) are found in the same locus in humans as they are in yeast (S. Cerevisiae) and the brown rat (R. Norvegicus).[9] This establishes the sequence conservation and importance of dyskerin within the eukaryotes. The relevant nature of dyskerin throughout most species is to catalyze the post-transcriptional pseudouridylation of specific uridines found in non-coding RNAs, such as ribosomal RNA (rRNA). Cbf5, the yeast analog of human dyskerin, is indeed known to be associated with the processing and maturation of rRNA.[6] In humans, this role can be attributed to dyskerin.[9] Thus, the X-linked form of this disease may result in specific issues related to dysfunctional RNA and perhaps a graver phenotype. Within the vertebrates, as opposed to single celled eukaryotes, dyskerin is a key component of the telomerase RNA component (TERC) in the form of the H/ACA motif.[10] This X-linked variety, like the Nop10 and Nhp2 mutations, demonstrates shortened telomeres as a result of lower TERC concentrations. [11]
Autosomal dominant
3 genes: TERC, TERT, TINF2 The evidence supporting the importance of the H/ACA domain in human telomerase is abundant. At least one study[12] has shown that these mutations affect telomerase activity by negatively affecting pre-RNP assembly and maturation of human telomerase RNA. Nonetheless, mutations that directly affect the telomerase RNA components would presumably exist and should also cause premature aging or DKC-like symptoms. Indeed, three families with mutations in the human TERC gene have been studied with intriguing results.[6] In two of these families, two family-specific single nucleotide polymorphisms were present while in the other there persisted a large-scale deletion (821 base pairs of DNA) on chromosome 3 which includes 74 bases coding for a section of the H/ACA domain. These three different mutations result in a mild form of dyskeratosis congenita which uniquely follows an autosomal dominant pattern of inheritance. Premature graying, early dental loss, predisposition to skin cancer, as well as shortening of telomere length continue to be characteristic of this disease.[13]
Autosomal recessive
6 genes: The true phenotype of DKC individuals may depend upon which protein has incurred a mutation. One documented autosomal recessive mutation[7] in a family that carries DKC has been found in NOP10. Specifically, the mutation is a change of base from cytosine to thymine in a highly conserved region of the NOP10 sequence. This mutation, on chromosome 15, results in an amino acid change from arginine to tryptophan. Homozygous recessive individuals show the symptoms of dyskeratosis congenita in full. As compared to age-matched normal individuals, those suffering from DKC have telomeres of a much shorter length. Furthermore, heterozygotes, those who have one normal allele and one coding for the disease, also show relatively shortened telomeres. The cause of this was determined to be a reduction in TERC levels in those with the Nop10 mutation. With TERC levels down, telomere maintenance, especially in development, would be presumed to suffer accordingly. This would lead to the telomere shortening described.[7]
NHP2 mutations are similar in characterization to NOP10. These mutations are also autosomal recessive with three specific single-nucleotide polymorphisms being recognized which result in dyskeratosis congenita. Also, like NOP10, individuals with these NHP2 mutations have a reduction in the amount of telomerase RNA component (TERC) present in the cell. Again, it can be presumed that a reduction in TERC results in aberrant telomere maintenance and thus shortened telomeres. Those homozygous recessive for mutations in NHP2 do show shorter telomeres when compared with age-matched normal individuals.[8]
Pathophysiology
Dyskeratosis congenita is a disorder of poor telomere maintenance[6] mainly due to a number of gene mutations that give rise to abnormal ribosome function, termed ribosomopathy. Specifically, the disease is related to one or more mutations which directly or indirectly affect the vertebrate telomerase RNA component (TERC). [14]
Telomerase is a reverse transcriptase which maintains a specific repeat sequence of DNA, the telomere, during development. Telomeres are placed by telomerase on both ends of linear chromosomes as a way to protect linear DNA from general forms of chemical damage and to correct for the chromosomal end-shortening that occurs during normal DNA replication.[15] This end-shortening is the result of the eukaryotic DNA polymerases having no mechanism for synthesizing the final nucleotides present on the end of the "lagging strand" of double stranded DNA. DNA polymerase can only synthesize new DNA from an old DNA strand in the 5'→3' direction. Given that DNA has two strands that are complementary, one strand must be 5'→3' while the other is 3'→5'. This inability to synthesize in the 3'→5' directionality is compensated with the use of Okazaki fragments, short pieces of DNA that are synthesized 5'→3' from the 3'→5' as the replication fork moves. As DNA polymerase requires RNA primers for DNA binding in order to commence replication, each Okazaki fragment is thus preceded by an RNA primer on the strand being synthesized. When the end of the chromosome is reached, the final RNA primer is placed upon this nucleotide region, and it is inevitably removed. Unfortunately once the primer is removed, DNA polymerase is unable to synthesize the remaining bases.[15][16]
Sufferers of DKC have been shown to have a reduction in TERC levels invariably affecting the normal function of telomerase which maintains these telomeres.[6][7][9] With TERC levels down, telomere maintenance during development suffers accordingly. In humans, telomerase is inactive in most cell types after early development (except in extreme cases such as cancer).[10] Thus, if telomerase is not able to efficiently affect the DNA in the beginning of life, chromosomal instability becomes a grave possibility in individuals much earlier than would be expected. [17]
A study shows that proliferative defects in DC skin keratinocytes are corrected by expression of the telomerase reverse transcriptase, TERT, or by activation of endogenous telomerase through expression of papillomavirus E6/E7 of the telomerase RNA component, TERC.[18]
Diagnosis
Since the disease has a wide variety of symptoms due to involvement of multiple systems of the body, diagnostic testing depends on the clinical findings in each individual patient. Commonly used tests include a complete blood count (CBC), bone marrow examination, leukocyte telomere length test (e.g. Flow FISH), pulmonary function test, and genetic testing.[19][20]
Management
The mainstay of treatment in dyskeratosis congenita is hematopoietic stem cell transplantation, best outcome with sibling donor. Short term therapy in initial stages is with anabolic steroids [oxymetholone, danazol] or with erythropoietin-like hormones or with granulocyte-colony stimulating factor [filgrastim) all these therapies are directed to cope with effects of bone marrow failure which manifests as low red and white blood cell counts. These medications help to increase the blood components and make up for the deficiencies caused due to bone marrow failure. Dyskeratosis Congenita in regards to stem cell transplantation have to be very carefully treated with low intensity radiation/chemo to avoid potentially catastrophic effects of Host versus graft disease and toxicity to other organs affected by short telomeres which makes them very sensitive to any radiation especially the lungs, and liver. [21]
Prognosis
DC is associated with shorter life expectancy, but many live to at least age 60.[22] Main cause of mortality in these patients are related to bone marrow failure. Nearly 80% of the patients of dyskeratosis congenita develop bone marrow failure.[23]
Research
Recent research has used induced pluripotent stem cells to study disease mechanisms in humans, and discovered that the reprogramming of somatic cells restores telomere elongation in dyskeratosis congenita (DKC) cells despite the genetic lesions that affect telomerase. The reprogrammed DKC cells were able to overcome a critical limitation in TERC levels and restored function (telomere maintenance and self-renewal). Therapeutically, methods aimed at increasing TERC expression could prove beneficial in DKC.[24]
References
- Online Mendelian Inheritance in Man (OMIM): 305000
- James W, Berger T, Elston D (2005). Andrews' Diseases of the Skin: Clinical Dermatology (10th ed.). Saunders. ISBN 0-7216-2921-0..
- Online Mendelian Inheritance in Man (OMIM): 127550
- Kirwan M, Dokal I (April 2009). "Dyskeratosis congenita, stem cells and telomeres". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1792 (4): 371–379. doi:10.1016/j.bbadis.2009.01.010. PMC 2686081. PMID 19419704.
- Peters JJ (1988). "Module # 4: Geriatric Syndromes" (PDF). Geriatrics, Palliative Care and Interprofessional Teamwork Curriculum. 4: 27. Archived (PDF) from the original on 2016-12-22. Retrieved 2022-08-20 – via New York and New Jersey VA Health Care Network.
- Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, Dokal I (September 2001). "The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita". Nature. 413 (6854): 432–435. Bibcode:2001Natur.413..432V. doi:10.1038/35096585. PMID 11574891. S2CID 4348062.
- Walne AJ, Vulliamy T, Marrone A, Beswick R, Kirwan M, Masunari Y, et al. (July 2007). "Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10". Human Molecular Genetics. 16 (13): 1619–1629. doi:10.1093/hmg/ddm111. PMC 2882227. PMID 17507419.
- Vulliamy T, Beswick R, Kirwan M, Marrone A, Digweed M, Walne A, Dokal I (June 2008). "Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita". Proceedings of the National Academy of Sciences of the United States of America. 105 (23): 8073–8078. doi:10.1073/pnas.0800042105. PMC 2430361. PMID 18523010.
- Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ, et al. (May 1998). "X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions". Nature Genetics. 19 (1): 32–38. doi:10.1038/ng0598-32. PMID 9590285. S2CID 205342127.
- Wong JM, Collins K (October 2006). "Telomerase RNA level limits telomere maintenance in X-linked dyskeratosis congenita". Genes & Development. 20 (20): 2848–2858. doi:10.1101/gad.1476206. PMC 1619937. PMID 17015423.
- Wong JM, Collins K (October 2006). "Telomerase RNA level limits telomere maintenance in X-linked dyskeratosis congenita". Genes & Development. 20 (20): 2848–2858. doi:10.1101/gad.1476206. PMC 1619937. PMID 17015423.
- Trahan C, Dragon F (February 2009). "Dyskeratosis congenita mutations in the H/ACA domain of human telomerase RNA affect its assembly into a pre-RNP". RNA. 15 (2): 235–243. doi:10.1261/rna.1354009. PMC 2648702. PMID 19095616.
- Kirwan M, Dokal I (April 2009). "Dyskeratosis congenita, stem cells and telomeres". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1792 (4): 371–379. doi:10.1016/j.bbadis.2009.01.010. PMC 2686081. PMID 19419704.
- Dorgaleleh S, Naghipoor K, Hajimohammadi Z, Dastaviz F, Oladnabi M (February 2022). "Molecular insight of dyskeratosis congenita: Defects in telomere length homeostasis". Journal of Clinical and Translational Research. 8 (1): 20–30. PMC 8791241. PMID 35097237.
- Greider CW (May 1996). "Telomere length regulation". Annual Review of Biochemistry. 65: 337–365. doi:10.1146/annurev.bi.65.070196.002005. PMID 8811183. S2CID 45169518.
- Watson JD, Baker TA, Bell SP, Gann A, Levine M, Losick RM (2004). Molecular Biology of the Gene (5th ed.). San Francisco: Pearson/Benjamin Cummings. OCLC 1256524135.
- Sarek G, Marzec P, Margalef P, Boulton SJ (November 2015). "Molecular basis of telomere dysfunction in human genetic diseases". Nature Structural & Molecular Biology. 22 (11): 867–874. doi:10.1038/nsmb.3093. PMID 26581521. S2CID 205523942.
- Gourronc FA, Robertson MM, Herrig AK, Lansdorp PM, Goldman FD, Klingelhutz AJ (March 2010). "Proliferative defects in dyskeratosis congenita skin keratinocytes are corrected by expression of the telomerase reverse transcriptase, TERT, or by activation of endogenous telomerase through expression of papillomavirus E6/E7 or the telomerase RNA component, TERC". Experimental Dermatology. 19 (3): 279–288. doi:10.1111/j.1600-0625.2009.00916.x. PMC 2852488. PMID 19558498.
- Fernández García MS, Teruya-Feldstein J (2014-08-21). "The diagnosis and treatment of dyskeratosis congenita: a review". Journal of Blood Medicine. 5: 157–167. doi:10.2147/JBM.S47437. PMC 4145822. PMID 25170286.
- Savage SA, Niewisch MR (2019-11-21). "Dyskeratosis Congenita and Related Telomere Biology Disorders". In Adam MP, Ardinger HH, Pagon RA, Wallace SE (eds.). GeneReviews. Seattle (WA): University of Washington, Seattle. PMID 20301779. Retrieved 2020-12-27.
- Fioredda F, Iacobelli S, Korthof ET, Knol C, van Biezen A, Bresters D, et al. (October 2018). "Outcome of haematopoietic stem cell transplantation in dyskeratosis congenita". British Journal of Haematology. 183 (1): 110–118. doi:10.1111/bjh.15495. PMID 29984823. S2CID 51602130.
- Alter BP, Rosenberg PS, Giri N, Baerlocher GM, Lansdorp PM, Savage SA (March 2012). "Telomere length is associated with disease severity and declines with age in dyskeratosis congenita". Haematologica. 97 (3): 353–359. doi:10.3324/haematol.2011.055269. PMC 3291588. PMID 22058220.
- Dokal I (10 December 2011). "Dyskeratosis congenita". Hematology. American Society of Hematology. Education Program. 2011 (1): 480–486. doi:10.1182/asheducation-2011.1.480. PMID 22160078.
- Agarwal S, Loh YH, McLoughlin EM, Huang J, Park IH, Miller JD, et al. (March 2010). "Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients". Nature. 464 (7286): 292–296. Bibcode:2010Natur.464..292A. doi:10.1038/nature08792. PMC 3058620. PMID 20164838.