Wiskott–Aldrich syndrome
Wiskott–Aldrich syndrome (WAS) is a rare X-linked recessive disease characterized by eczema, thrombocytopenia (low platelet count), immune deficiency, and bloody diarrhea (secondary to the thrombocytopenia).[1] It is also sometimes called the eczema-thrombocytopenia-immunodeficiency syndrome in keeping with Aldrich's original description in 1954.[2] The WAS-related disorders of X-linked thrombocytopenia (XLT) and X-linked congenital neutropenia (XLN) may present with similar but less severe symptoms and are caused by mutations of the same gene.
| Wiskott-Aldrich syndrome | |
|---|---|
 | |
| A) Multiple face petechiae and a hematoma under the right eye (left in image). B) Eczema of the foot. | |
| Specialty | Immunology |
Signs and symptoms
WAS occurs most often in males due to its X-linked recessive pattern of inheritance, affecting between 1 and 10 males per million.[1] The first signs are usually petechiae and bruising, resulting from a low platelet count (i.e. thrombocytopenia). Spontaneous nose bleeds and bloody diarrhea are also common and eczema typically develops within the first month of life. Recurrent bacterial infections typically develop by three months of age. The majority of children with WAS develop at least one autoimmune disorder, and cancers (mainly lymphoma and leukemia) develop in up to a third of patients.[3] Immunoglobulin M (IgM) levels are reduced, IgA and IgE are elevated, and IgG levels can be normal, reduced, or elevated.[4] In addition to thrombocytopenia, WAS patients have abnormally small platelets (i.e. microthrombocytes) and ~30% also have elevated eosinophil counts (i.e. eosinophilia).[5]
Pathophysiology
The microthrombocytes seen in WAS patients have only been observed in one other condition, ARPC1B deficiency.[6] In both conditions the defective platelets are thought to be removed from circulation by the spleen and/or liver, leading to low platelet counts. WAS patients have increased susceptibility to infections, particularly of the ears and sinuses, and this immune deficiency has been linked to decreased antibody production and the inability of immune T cells to effectively combat infection.[7]
Genetics
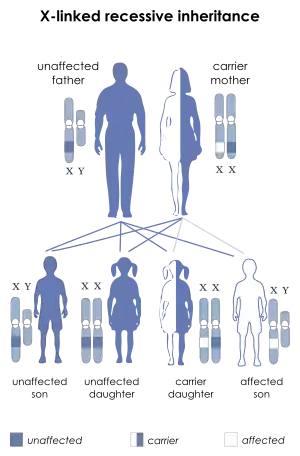
WAS is associated with mutations in a gene on the short arm of the X chromosome (Xp11.23) that was originally termed the Wiskott-Aldrich syndrome protein gene and is officially known as WAS (Gene ID: 7454).[8] X-linked thrombocytopenia (XLT) is also linked to pathogenic variants in the WAS gene, although some variants tend to be more strongly associated with XLT versus others that are more associated with WAS. The rare disorder X-linked neutropenia has also been linked to a specific subset of WAS mutations.[9]
The protein product of WAS is known as WASp. It contains 502 amino acids and is mainly expressed in hematopoietic cells (the cells in the bone marrow that develop into blood cells). The main function of WASp is to activate actin polymerization by serving as a nucleation-promoting factor (NPF) for the Arp2/3 complex, which generates branched actin filaments. Several proteins can serve as NPFs, and it has been observed that in WAS platelets the Arp2/3 complex functions normally, indicating that WASp is not required for its activation in platelets.[10] In T-cells, WASp is important because it is known to be activated via T-cell receptor signaling pathways to induce cortical actin cytoskeleton rearrangements that are responsible for forming the immunological synapse.[11]
The severity of the symptoms produced by pathogenic variants in the WAS gene generally correlates with their effects on WASp. Missense variants generally are associated with less severe disease than truncating variants that produce no protein due to nonsense-mediated decay.[12] However, this correlation is not perfect, and sometimes the same variant can be seen both in XLT and in WAS (sometimes within two different members of the same family), a concept in genetics referred to as variable expressivity.[13] Although autoimmune disease and malignancy may occur in both conditions, patients with loss of WASp are at higher risk. A defect in the CD43 molecule has also been found in WAS patients.[14] CD43, a transmembrane sialoglycoprotein also known as a leukosialin, is part of a greater complex involved in T-cell activation and acts as a sensitive indicator of abnormal, malignant B cell populations.[15] Defects in this molecule may be detrimental to WAS patients, who are at a much higher risk of autoimmune diseases that may be exasperated in later-detected B-cell lymphomas.
Diagnosis
The diagnosis can be made on the basis of clinical findings, the peripheral blood smear, and low immunoglobulin levels. Typically, IgM levels are low, IgA levels are elevated, and IgE levels may be elevated; paraproteins are occasionally observed.[16] Skin immunologic testing (allergy testing) may reveal hyposensitivity. Individuals with Wiskott–Aldrich syndrome however are at higher risk for severe food allergies.[17] Not all patients have a positive family history of the disorder; new mutations do occur. Often, leukemia may be suspected on the basis of low platelets and infections, and bone marrow biopsy may be performed. Decreased levels of WASp are typically observed. The current gold standard for diagnosis is DNA sequence analysis, which can detect WAS and the related disorders XLT and XLN in 95% of patients and carriers.[18]
| Mandatory criteria |
|
| Definitive |
|
| Probable |
|
| Possible |
|
Classification
Jin et al. (2004) employ a numerical grading of severity:[12] This score, which ranges from 0 to 5, may have clinical utility for predicting disease severity.[21] Those with higher WAS scores (e.g., 5) at younger ages (e.g., age less than 5 years old), are thought to be at highest risk for increased morbidity and mortality related to their condition.[22] As individuals can develop more WAS-related symptoms (e.g. autoimmune disease, malignancy) with age, one's WAS score can increase over time. A lower WAS score may be more compatible with conservative management versus higher WAS scores that may favor intervention with treatments such as hematopoietic stem cell transplant.
| Score | Definition | Clinical syndrome |
|---|---|---|
| 0 | Neutropenia (low white blood cell count) or myelodysplasia only | X-linked neutropenia (XLN) |
| 0.5 | Intermittent thrombocytopenia (low platelet counts sometimes but not always) | X-linked thrombocytopenia (XLT) |
| 1 | Thrombocytopenia and small platelets (microthrombocytopenia) | XLT |
| 2 | Microthrombocytopenia plus normally responsive eczema or occasional upper respiratory tract infections | XLT |
| 2.5 | Microthrombocytopenia plus therapy-responsive but severe eczema or airway infections requiring antibiotics | XLT/Wiskott–Aldrich syndrome (WAS) |
| 3 | Microthrombocytopenia plus both eczema and airway infections requiring antibiotics | WAS |
| 4 | Microthrombocytopenia plus eczema continuously requiring therapy and/or severe or life-threatening infections | WAS |
| 5 | Microthrombocytopenia plus autoimmune disease or malignancy | XLT/WAS + autoimmune disease or cancer |
Treatment
Hematopoietic stem cell transplant
Treatment of Wiskott–Aldrich syndrome depends on the severity of the disease. WAS is primarily a disorder of the blood-forming tissues, so in cases of severe disease (WAS score 3–5) the only widely available curative treatment currently available is a hematopoietic stem cell transplant (HCT). In this procedure stem cells are harvested from umbilical cord blood, bone marrow, or peripheral blood following treatment with medications that cause stem cells to leave the bone marrow and circulate systemically. The best outcomes are with HLA-identical or similar donors (often siblings). In cases of milder disease the potential benefits of HCT (>90% probability of cure if transplant occurs before age two) must be considered in the context of non-trivial risks presented by the procedure itself and the potential need for lifelong immunosuppression to prevent graft-versus-host disease.[23][24] Generally outcomes are better if HCT occurs prior to the development of autoimmune disease or malignancy, however there are risks associated with chemotherapy (needed to make room for the new stem cells) especially in young infants (risk of a second cancer, or infertility).
Bleeding complications
Otherwise WAS treatment is focused on managing symptoms and preventing complications. The greatest mortality risk in WAS before age 30 is from bleeding so aspirin and other nonsteroidal anti-inflammatory drugs that may interfere with already compromised platelet function should generally be avoided.[13] Circumcision, as well as elective surgeries, should generally be deferred in males with thrombocytopenia until after HCT if possible. Protective helmets can help protect children from life-threatening intracranial hemorrhage (brain bleed) which could result from head injuries. Patients may require platelet transfusions when there is extreme bloodloss (such as during surgery) or for very low platelets splenectomy (removal of the spleen) may also be lifesaving.[25] However, splenectomy is generally considered palliative and is not universally recommended in WAS because it can increase the risk of life-threatening infections.[26][13] Post-splenectomy patients will require lifelong antibiotic prophyllaxis to prevent infections. Study of eltrombopag, a thrombopoietic agent used to increase platelets in immune thrombocytopenic purpura (ITP), in WAS concluded that although it increased platelet numbers it failed to increase platelet activation in most patients.[27] It has since been proposed the eltrombopag may be used to bridge to HCT in patients with severe thrombocytopenia to normalize platelet numbers without transfusions and decrease bleeding events.[28] Anemia from bleeding may require iron supplementation or blood transfusion. Regular surveillance of blood counts is recommended.
Infections and autoimmune disease
For patients with frequent infections, intravenous immunoglobulins (IVIG) or subcutaneous immunoglobulins can be regularly scheduled to boost the immune system. Adequacy of IVIG replacement can be assessed via periodic lab draws. WAS patients with immune system compromise may benefit from antibiotic prophylaxis, for example by taking trimethoprim-sulfamethoxazole to prevent Pneumocystis jirovecii-related pneumonia. Similarly, prophylactic antibiotic use may also be considered in patients with recurrent bacterial sinus or lung infections. When there are signs or symptoms of an infection, prompt and thorough evaluation is important including blood cultures to guide therapy (often IV antibiotics). Live vaccines (such as MMR or rotavirus) should be avoided during routine childhood vaccination. Inactivated vaccines may be given safely but may not provide protective levels of immunity. Eczema is generally treated with topical steroids, and if chronic skin infections exacerbate eczema an antibiotic may also be given. Autoimmune disease is managed with judicious use of appropriate immunosuppressants.
Gene therapy
For severely affected males without an HLA-matched donor, studies of correcting Wiskott–Aldrich syndrome with gene therapy using a lentivirus are underway.[29][30] Proof-of-principle for successful hematopoietic stem cell gene therapy has been provided for patients with Wiskott–Aldrich syndrome.[31] In July 2013 the Italian San Raffaele Telethon Institute for Gene Therapy (HSR-TIGET) reported that three children with Wiskott–Aldrich syndrome showed significant improvement (improved platelet counts, immune functiona, and clinical symptoms) 20–30 months after being treated with a genetically modified lentivirus.[32] In April 2015 results from a follow-up British and French trial six out of seven individuals showed improvement of immune function and clinical symptoms an average of 27 months after treatment with gene therapy.[33][34][35] Importantly, neither study showed evidence of leukemic proliferation following treatment, a complication of early attempts at gene therapy using a retroviral vector.[36] It is unknown why these gene therapies did not restore normal platelet numbers, but gene therapy treatment was still associated with transfusion-independence and a significant reduction in bleeding events.[32][33] A version of this treatment, OTL-103, is being developed by Orchard Therapeutics and (as of 28 June 2021) is undergoing Phase I/II clinical trials.
Prognosis
Outcomes from Wiskott–Aldrich syndrome are variable and depend on how severely an individual is affected (the WAS score may be used to assess disease severity). The milder end of the disease spectrum associated with the WAS gene is referred to as X-linked neutropenia or X-linked thrombocytopenia, and the latter is thought to have a normal life expectancy with reports of minimally affected males surviving into their seventh decade without treatment.[21] Traditionally however Wiskott–Aldrich syndrome has been associated with premature death from causes including bleeding, infections, or malignancy.[37] Wiskott–Aldrich syndrome is a condition with variable expressivity, meaning that even within the same family some may exhibit only chronic thrombocytopenia while others experience severe, life-threatening complications of Wiskott–Aldrich syndrome in infancy or childhood.[38][13] Given symptoms often progress with age, it is challenging to predict how affected a newly diagnosed infant will eventually be. There is some genotype-phenotype correlation, with most individuals with X-linked thrombocytopenia having missense variants in the WAS gene versus 86.5% of those that make no WAS protein having the classic Wiskott–Aldrich syndrome phenotype.[39][40] Overall the prognosis for individuals with Wiskott–Aldrich syndrome has improved considerably over the past decades due to earlier diagnoses and more access to treatments.
Epidemiology
The estimated incidence of Wiskott–Aldrich syndrome in the United States is one in 250,000 live male births.[41] While still a rare disease, this makes it more common than many genetic immunodeficiency syndromes such as hyper-IgM syndrome or SCID, which have an estimated incidence of about one in 1,000,000 live births, and Wiskott–Aldrich syndrome is thought to account for 1.2% of all inherited immunodeficiencies in the United States.[42] WAS occurs worldwide and is not known to be more common in any particular ethnic group.
History
The syndrome is named after Dr. Alfred Wiskott (1898–1978), a German pediatrician who first noticed the syndrome in 1937,[43] and Dr. Robert Anderson Aldrich (1917–1998), an American pediatrician who described the disease in a family of Dutch-Americans in 1954.[2] Wiskott described three brothers with a similar disease, whose sisters were unaffected. In 2006, a German research group analyzed family members of Wiskott's three cases, and surmised they probably shared a novel frameshift mutation of the first exon of the WASp gene.[44]
References
- "Wiskott-Aldrich syndrome". Genetics Home Reference. Retrieved 2016-06-26.
- Aldrich RA, Steinberg AG, Campbell DC (February 1954). "Pedigree demonstrating a sex-linked recessive condition characterized by draining ears, eczematoid dermatitis and bloody diarrhea". Pediatrics. 13 (2): 133–9. doi:10.1542/peds.13.2.133. PMID 13133561. S2CID 42440727.
- Wiskott-Aldrich Syndrome at eMedicine
- Sande MA, Wilson WP (2001). Current diagnosis & treatment in infectious diseases. New York: Lange Medical Books/McGraw-Hill. p. 361. ISBN 978-0-8385-1494-8.
- Navabi B, Upton JE (2016). "Primary immunodeficiencies associated with eosinophilia". Allergy, Asthma, and Clinical Immunology. 12: 27. doi:10.1186/s13223-016-0130-4. PMC 4878059. PMID 27222657.
- Kahr WH, Pluthero FG, Elkadri A, Warner N, Drobac M, Chen CH, et al. (April 2017). "Loss of the Arp2/3 complex component ARPC1B causes platelet abnormalities and predisposes to inflammatory disease". Nature Communications. 8: 14816. Bibcode:2017NatCo...814816K. doi:10.1038/ncomms14816. PMC 5382316. PMID 28368018.
- "Wiskott-Aldrich Syndrome: Immunodeficiency Disorders: Merck Manual Professional". Retrieved 2008-03-01.
- Derry JM, Ochs HD, Francke U (August 1994). "Isolation of a novel gene mutated in Wiskott-Aldrich syndrome". Cell. 78 (4): 635–44. doi:10.1016/0092-8674(94)90528-2. PMID 8069912. S2CID 9040376.
- Westerberg LS, Meelu P, Baptista M, Eston MA, Adamovich DA, Cotta-de-Almeida V, et al. (June 2010). "Activating WASP mutations associated with X-linked neutropenia result in enhanced actin polymerization, altered cytoskeletal responses, and genomic instability in lymphocytes". The Journal of Experimental Medicine. 207 (6): 1145–52. doi:10.1084/jem.20091245. PMC 2882832. PMID 20513746.
- Falet H, Hoffmeister KM, Neujahr R, Hartwig JH (September 2002). "Normal Arp2/3 complex activation in platelets lacking WASp". Blood. 100 (6): 2113–22. doi:10.1182/blood.V100.6.2113. PMID 12200375.
- Malinova D, Fritzsche M, Nowosad CR, Armer H, Munro PM, Blundell MP, et al. (May 2016). "WASp-dependent actin cytoskeleton stability at the dendritic cell immunological synapse is required for extensive, functional T cell contacts". Journal of Leukocyte Biology. 99 (5): 699–710. doi:10.1189/jlb.2a0215-050rr. PMC 5404712. PMID 26590149.
- Jin Y, Mazza C, Christie JR, Giliani S, Fiorini M, Mella P, et al. (December 2004). "Mutations of the Wiskott-Aldrich Syndrome Protein (WASP): hotspots, effect on transcription, and translation and phenotype/genotype correlation". Blood. 104 (13): 4010–9. doi:10.1182/blood-2003-05-1592. PMID 15284122.
- Albert MH, Notarangelo LD, Ochs HD (January 2011). "Clinical spectrum, pathophysiology and treatment of the Wiskott-Aldrich syndrome". Current Opinion in Hematology. 18 (1): 42–8. doi:10.1097/moh.0b013e32834114bc. PMID 21076297. S2CID 459396.
- Rosenstein Y, Park JK, Hahn WC, Rosen FS, Bierer BE, Burakoff SJ (November 1991). "CD43, a molecule defective in Wiskott-Aldrich syndrome, binds ICAM-1". Nature. 354 (6350): 233–5. Bibcode:1991Natur.354..233R. doi:10.1038/354233a0. PMID 1683685. S2CID 4256329.
- "CD43 - an overview | ScienceDirect Topics". www.sciencedirect.com. Retrieved 2021-11-19.
- Radl J, Dooren LH, Morell A, Skvaril F, Vossen JM, Uittenbogaart CH (August 1976). "Immunoglobulins and transient paraproteins in sera of patients with the Wiskott-Aldrich syndrome: a follow-up study". Clinical and Experimental Immunology. 25 (2): 256–63. PMC 1541349. PMID 954233.
- Liang Y, Gudjonsson JE (October 2016). "WASP, Tregs, and food allergies - rare disease provides insight into a common problem". The Journal of Clinical Investigation. 126 (10): 3728–3730. doi:10.1172/JCI90198. PMC 5096819. PMID 27643436.
- Chandra S, Bronicki L, Nagaraj CB, Zhang K (1993). "WAS-Related Disorders". In Adam MP, Ardinger HH, Pagon RA, Wallace SE (eds.). GeneReviews. University of Washington, Seattle. PMID 20301357. Retrieved 2020-12-10.
- "ESID - European Society for Immunodeficiencies". esid.org. Retrieved 2020-12-10.
- Catucci M, Castiello MC, Pala F, Bosticardo M, Villa A (2012). "Autoimmunity in wiskott-Aldrich syndrome: an unsolved enigma". Frontiers in Immunology. 3: 209. doi:10.3389/fimmu.2012.00209. PMC 3399097. PMID 22826711.
- Albert MH, Bittner TC, Nonoyama S, Notarangelo LD, Burns S, Imai K, et al. (April 2010). "X-linked thrombocytopenia (XLT) due to WAS mutations: clinical characteristics, long-term outcome, and treatment options". Blood. 115 (16): 3231–8. doi:10.1182/blood-2009-09-239087. PMID 20173115.
- Mahlaoui N, Pellier I, Mignot C, Jais JP, Bilhou-Nabéra C, Moshous D, et al. (February 2013). "Characteristics and outcome of early-onset, severe forms of Wiskott-Aldrich syndrome". Blood. 121 (9): 1510–6. doi:10.1182/blood-2012-08-448118. PMID 23264593.
- Moratto, Daniele; Giliani, Silvia; Bonfim, Carmem; Mazzolari, Evelina; Fischer, Alain; Ochs, Hans D.; Cant, Andrew J.; Thrasher, Adrian J.; Cowan, Morton J.; Albert, Michael H.; Small, Trudy (2011-08-11). "Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott-Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980-2009: an international collaborative study". Blood. 118 (6): 1675–1684. doi:10.1182/blood-2010-11-319376. ISSN 0006-4971. PMC 3156052. PMID 21659547.
- Shin, C R; Kim, M-O; Li, D; Bleesing, J J; Harris, R; Mehta, P; Jodele, S; Jordan, M B; Marsh, R A; Davies, S M; Filipovich, A H (2012-03-19). "Outcomes following hematopoietic cell transplantation for Wiskott–Aldrich syndrome". Bone Marrow Transplantation. 47 (11): 1428–1435. doi:10.1038/bmt.2012.31. ISSN 0268-3369. PMID 22426750. S2CID 13401119.
- Mullen, CA; Anderson, KD; Blaese, RM (1993-11-15). "Splenectomy and/or bone marrow transplantation in the management of the Wiskott-Aldrich syndrome: long-term follow-up of 62 cases". Blood. 82 (10): 2961–2966. doi:10.1182/blood.v82.10.2961.bloodjournal82102961. ISSN 0006-4971. PMID 8219187.
- Ozsahin, Hulya; Cavazzana-Calvo, Marina; Notarangelo, Luigi D.; Schulz, Ansgar; Thrasher, Adrian J.; Mazzolari, Evelina; Slatter, Mary A.; Le Deist, Francoise; Blanche, Stephane; Veys, Paul; Fasth, Anders (2008-01-01). "Long-term outcome following hematopoietic stem-cell transplantation in Wiskott-Aldrich syndrome: collaborative study of the European Society for Immunodeficiencies and European Group for Blood and Marrow Transplantation". Blood. 111 (1): 439–445. doi:10.1182/blood-2007-03-076679. ISSN 0006-4971. PMID 17901250. S2CID 10492644.
- Gerrits, Anja J.; Leven, Emily A.; Frelinger, Andrew L.; Brigstocke, Sophie L.; Berny-Lang, Michelle A.; Mitchell, W. Beau; Revel-Vilk, Shoshana; Tamary, Hannah; Carmichael, Sabrina L.; Barnard, Marc R.; Michelson, Alan D. (2015-09-10). "Effects of eltrombopag on platelet count and platelet activation in Wiskott-Aldrich syndrome/X-linked thrombocytopenia". Blood. 126 (11): 1367–1378. doi:10.1182/blood-2014-09-602573. ISSN 0006-4971. PMC 4729539. PMID 26224646.
- Gabelli, Maria; Marzollo, Antonio; Notarangelo, Lucia Dora; Basso, Giuseppe; Putti, Maria Caterina (December 2017). "Eltrombopag use in a patient with Wiskott-Aldrich syndrome". Pediatric Blood & Cancer. 64 (12): e26692. doi:10.1002/pbc.26692. PMID 28643468. S2CID 44479925.
- Galy A, Roncarolo MG, Thrasher AJ (February 2008). "Development of lentiviral gene therapy for Wiskott Aldrich syndrome". Expert Opinion on Biological Therapy. 8 (2): 181–90. doi:10.1517/14712598.8.2.181. PMC 2789278. PMID 18194074.
- Frecha C, Toscano MG, Costa C, Saez-Lara MJ, Cosset FL, Verhoeyen E, Martin F (June 2008). "Improved lentiviral vectors for Wiskott-Aldrich syndrome gene therapy mimic endogenous expression profiles throughout haematopoiesis". Gene Therapy. 15 (12): 930–41. doi:10.1038/gt.2008.20. PMID 18323794.
- Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Díez IA, Dewey RA, et al. (November 2010). "Stem-cell gene therapy for the Wiskott-Aldrich syndrome". The New England Journal of Medicine. 363 (20): 1918–27. doi:10.1056/NEJMoa1003548. PMC 3064520. PMID 21067383.
- Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, et al. (August 2013). "Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome". Science. 341 (6148): 1233151. doi:10.1126/science.1233151. PMC 4375961. PMID 23845947.
- Hacein-Bey Abina, Salima; Gaspar, H. Bobby; Blondeau, Johanna; Caccavelli, Laure; Charrier, Sabine; Buckland, Karen; Picard, Capucine; Six, Emmanuelle; Himoudi, Nourredine; Gilmour, Kimberly; McNicol, Anne-Marie (2015-04-21). "Outcomes Following Gene Therapy in Patients With Severe Wiskott-Aldrich Syndrome". JAMA. 313 (15): 1550–63. doi:10.1001/jama.2015.3253. ISSN 0098-7484. PMC 4942841. PMID 25898053.
- Gallagher, James (21 April 2015) Gene therapy: 'Tame HIV' used to cure disease BBC News, Health, Retrieved 21 April 2015
- Malech HL, Ochs HD (April 2015). "An emerging era of clinical benefit from gene therapy". JAMA. 313 (15): 1522–3. doi:10.1001/jama.2015.2055. PMID 25898049.
- Modlich, U; Schambach, A; Brugman, M H; Wicke, D C; Knoess, S; Li, Z; Maetzig, T; Rudolph, C; Schlegelberger, B; Baum, C (2008-05-22). "Leukemia induction after a single retroviral vector insertion in Evi1 or Prdm16". Leukemia. 22 (8): 1519–1528. doi:10.1038/leu.2008.118. ISSN 0887-6924. PMID 18496560. S2CID 20427830.
- Paller AS (May 1995). "A multiinstitutional survey of the Wiskott-Aldrich syndrome". Journal of the American Academy of Dermatology. 32 (5): 780–781. doi:10.1016/0190-9622(95)91469-2. ISSN 0190-9622.
- Buchbinder D, Nadeau K, Nugent D (October 2011). "Monozygotic twin pair showing discordant phenotype for X-linked thrombocytopenia and Wiskott-Aldrich syndrome: a role for epigenetics?". Journal of Clinical Immunology. 31 (5): 773–7. doi:10.1007/s10875-011-9561-3. PMID 21710275. S2CID 6820058.
- Imai K, Nonoyama S, Ochs HD (December 2003). "WASP (Wiskott-Aldrich syndrome protein) gene mutations and phenotype". Current Opinion in Allergy and Clinical Immunology. 3 (6): 427–36. doi:10.1097/00130832-200312000-00003. PMID 14612666. S2CID 11924967.
- Lutskiy MI, Rosen FS, Remold-O'Donnell E (July 2005). "Genotype-proteotype linkage in the Wiskott-Aldrich syndrome". Journal of Immunology. 175 (2): 1329–36. doi:10.4049/jimmunol.175.2.1329. PMID 16002738. S2CID 41185536.
- Robert A Schwartz, MD, MPH; Chief Editor: Harumi Jyonouchi, MD. Pediatric Wiskott-Aldrich Syndrome
- Buchbinder, David; Nugent, Diane; Fillipovich, Alexandra (April 2014). "Wiskott–Aldrich syndrome: diagnosis, current management, and emerging treatments". The Application of Clinical Genetics. 7: 55–66. doi:10.2147/tacg.s58444. ISSN 1178-704X. PMC 4012343. PMID 24817816.
- Wiskott A (1937). "Familiärer, angeborener Morbus Werlhofii? ("Familial congenital Werlhof's disease?")". Montsschr Kinderheilkd. 68: 212–16.
- Binder V, Albert MH, Kabus M, Bertone M, Meindl A, Belohradsky BH (October 2006). "The genotype of the original Wiskott phenotype". The New England Journal of Medicine. 355 (17): 1790–3. doi:10.1056/NEJMoa062520. PMID 17065640. S2CID 30040802.