Severe congenital neutropenia
Severe congenital neutropenia (SCN), also often known as Kostmann syndrome or disease, is a group of rare disorders that affect myelopoiesis, causing a congenital form of neutropenia, usually without other physical malformations. SCN manifests in infancy with life-threatening bacterial infections.[2]
| Severe congenital neutropenia | |
|---|---|
| Other names | Kostmann disease, Kostmann's agranulocytosis, Kostmann's syndrome, congenital agranulocytosis, congenital neutropenia, permanent neutropenia, infantile genetic agranulocytosis, severe infantile genetic neutropenia |
| Specialty | Hematology |
| Usual onset | Infancy[1] |
| Types | SCN1-SCN5, SCNX |
| Causes | Mutation in genes, depending on type[1] |
| Diagnostic method | Blood test, genetic testing[1] |
| Treatment | G-CSF, HSCT[1] |
| Medication | Filgrastim[1] |
| Frequency | 2-3 in million (2018)[1] |
Most cases of SCN respond to treatment with granulocyte colony-stimulating factor (filgrastim), which increases the neutrophil count and decreases the severity and frequency of infections.[2] Although this treatment has significantly improved survival, people with SCN are at risk of long-term complications such as hematopoietic clonal disorders (myelodysplastic syndrome, acute myeloid leukemia).
Kostmann disease (SCN3), the initial subtype recognized, was clinically described in 1956. This type has an autosomal recessive inheritance pattern, whereas the most common subtype, SCN1, shows autosomal dominant inheritance.
Presentation
Infants with SCN have frequent infections: 50% have a significant infection within 1 month, most others by 6 months.[3] Their etiology is usually bacterial, especially staphylococcal, and they commonly involve abscesses, both cutaneous and of internal organs, pneumonia, mastoiditis (inflammation of the mastoid process), and sepsis. All of these are life-threatening for infants.[4]
Genetics
| OMIM | Name | Gene | Chromosome | Gene/Locus MIM number |
|---|---|---|---|---|
| 202700 | SCN1 | ELANE | 19p13.3 | 130130[5] |
| 613107 | SCN2 | GFI1 | 1p22.1 | |
| 610738 | SCN3 | HAX1 | 1q21.3 | |
| 612541 | SCN4 | G6PC3 | 17q21.31 | |
| 615285 | SCN5 | VPS45 | 1q21.2 | |
| 300299 | SCNX | WASP | Xp11.23 | 300392[6] |
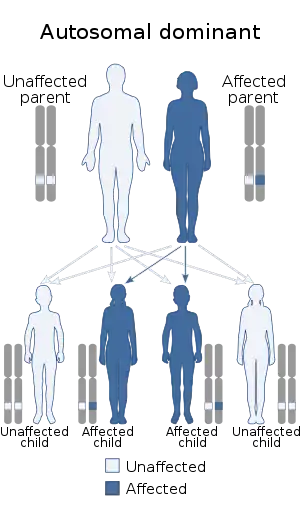

Kostmann disease, SCN3, is inherited in an autosomal recessive manner, but the commonest subtype of Kostmann syndrome, SCN1, is autosomal dominant.[7]
A significant proportion of SCN lacks a known mutation.[8] The recognized subtypes of Kostmann syndrome are:
- SCN1 is the commonest form of SCN, which accounts for 60-80% of SCN,[3] and the first to be genetically typified.[3] This autosomal dominant form that arises from mutations of the ELANE (formerly ELA2) gene on chromosome 19p13.3, which encodes neutrophil elastase.[5] Over a hundred ELANE mutations have been found in SCN1.[9] This same gene is mutated in cyclic neutropenia.[9]
- SCN2 is caused by heterozygous (autosomal dominant) mutation of the GFI1 gene on chromosome 1p22.[10] GFI1 is a repressor of several transcriptional processes, including ELANE,[3][10] as well as miR-21 and miR-196b micro-RNAs which influence myelopoiesis.[3]
- SCN3 is the "classical", autosomal recessive form of Kostmann disease which arises from homozygous mutations in the HAX1 gene on chromosome 1p22.1. About one third of SCN3 individuals also have neurological changes including seizures, learning disabilities, or developmental delay.[3]
- SCN4 is caused by autosomal recessive mutation of the G6PC3 gene on 17q21.[11] SCN4 is associated with structural cardiac abnormalities, enlarged liver, intermittent thrombocytopenia and a prominent superficial venous pattern.[11] A subset of SCN4 has severe primary pulmonary hypertension and respiratory failure.[11]
- SCN5 arises from autosomal recessive Thr224Asn mutation in the VPS45 gene on chromosome 1q21.2.[12] Unlike classical Kostmann disease, SCN5 also has defective platelet aggregation (thrombasthenia) and myelofibrosis.[13] This type is refractory to granulocyte colony-stimulating factor. There is an absence of lysosomes in fibroblasts and depletion of alpha granules in platelets. Accelerated apoptosis occurs in the neutrophils and bone marrow.
- X-linked SCN (SCNX) is caused by mutation in the WASP gene on Xp11.[14]
SCN occasionally may arise from SBDS mutations.[8]
Usage
Severe congenital neutropenia (SCN) is used as the overarching term for all diseases that affect myelopoiesis most prominently. Kostmann syndrome can restrictively refer to Kostmann disease specifically, or can be used synonymously with SCN as an umbrella term. These syndrome subtypes are phenotypically similar despite arising from different gene abnormalities.[3]
Kostmann disease is a form of severe congenital neutropenia (SCN), specifically type 3 (SCN3),[15] which is a rare autosomal recessive condition in which severe chronic neutropenia is detected soon after birth.[7][16] The disorder was discovered in 1956 in an extended family in northern Sweden by Rolf Kostmann, a Swedish doctor.[17][18]
Although mutations of more than 15 genes cause severe congenital neutropenia (in a general sense)[19] not all of these are usually considered as SCN. Clinical usage excludes two broad categories of congenital neutropenia. Diseases are excluded that overtly affect multiple systems rather than impacting myelopoiesis most prominently. Thus SCN excludes the severe neutropenia which can occur in congenital diseases such as Shwachman–Diamond syndrome, Barth syndrome, Chédiak–Higashi syndrome, WHIM syndrome, and glycogen storage disease type Ib.[19] A further group of other miscellaneous inherited disorders, such as hyper IgM syndrome, Hermansky–Pudlak syndrome (HPS), Griscelli syndrome (GS), PN, P14 deficiency, Cohen syndrome, Charcot–Marie–Tooth disease (CMT) can show congenital neutropenia, but lack bone marrow findings typical of SCN.
This group of diseases may also have additional features such as partial albinism, retinopathy, or neuropathy, and are not inclined to degenerate into acute myelogenous leukemia.[3]
GATA2 deficiency
GATA2 deficiency is a grouping of several disorders caused by common defect, viz., familial or sporadic inactivating mutations in one of the two parental GATA2 genes. These autosomal dominant mutations cause a reduction, i.e. a haploinsufficiency, in the cellular levels of the gene's product, GATA2. The GATA2 protein is a transcription factor critical for the embryonic development, maintenance, and functionality of blood-forming, lympathic-forming, and other tissue-forming stem cells. In consequence of these mutations, cellular levels of GATA2 are deficient and individuals develop over time hematological, immunological, lymphatic, or other presentations that may begin as apparently benign abnormalities but commonly progress to a more serious disorder. A small but significant percentage of individuals with GATA2 deficiency's present with congenital neutropenia. This neutropenia is typically mild, often persists for years, and therefore is not a Kostmann syndrome disorder. Over time, however, the deficiency commonly progresses to include thrombocytopenia, increases susceptibility to infections due to, e.g. atypical mycobacteria or human papillomavirus, dysfunction of non-hematological organs, the myelodysplastic syndrome, and/or a leukemia, particularly acute myelogenous leukemia.[20][21][22]
Pathophysiology
The various mutations are responsible for the untimely initiation of apoptosis in myelocytes, usually at the promyelocyte stage, leading to their premature destruction or maturation arrest in the bone marrow.[23] The ineffective production of neutrophils leads to a decrease in the absolute neutrophil count and a subsequent increased susceptibility to infections. There may be, in addition, other underlying molecular/genetic changes producing DNA mutations and genome instability, which contribute to initiation and progression of this disease.
Diagnosis
An absolute neutrophil count (ANC) chronically less than 500/mm3, usually less than 200/mm3, is the main sign of Kostmann's. Other elements include the severity of neutropenia, the chronology (from birth; not emerging later), and other normal findings (hemoglobin, platelets, general body health). Isolated neutropenia in infants can occur in viral infections, autoimmune neutropenia of infancy, bone marrow suppression from a drug or toxin, hypersplenism, and passive placental transfer of maternal IgG.[3]
A bone marrow test can assist in diagnosis. The bone marrow usually shows early granulocyte precursors, but myelopoietic development stops ("arrests") at the promyelocyte and/or myelocyte stage, so that few maturing forms are seen. Neutrophil survival is normal.
Treatment
Regular administration of exogenous granulocyte colony-stimulating factor (filgrastim) clinically improves neutrophil counts and immune function and is the mainstay of therapy, although this may increase risk for myelofibrosis and acute myeloid leukemia in the long term.[24]
Over 90% of SCN responds to treatment with granulocyte colony-stimulating factor (filgrastim), which has significantly improved survival.
References
- Dale DC, Makaryan V (2018) [2002]. "ELANE-Related Neutropenia". GeneReviews. PMID 20301705. Retrieved 2019-08-12.
- Boztug, K; Appaswamy, G; Ashikov, A; Schäffer, AA; Salzer, U; Diestelhorst, J; Germeshausen, M; Brandes, G; Lee-Gossler, J; Noyan, F; Gatzke, AK; Minkov, M; Greil, J; Kratz, C; Petropoulou, T; Pellier, I; Bellanné-Chantelot, C; Rezaei, N; Mönkemöller, K; Irani-Hakimeh, N; Bakker, H; Gerardy-Schahn, R; Zeidler, C; Grimbacher, B; Welte, K; Klein, C (1 January 2009). "A syndrome with congenital neutropenia and mutations in G6PC3". The New England Journal of Medicine. 360 (1): 32–43. doi:10.1056/nejmoa0805051. PMC 2778311. PMID 19118303.
- Hoffman, R; Benz, EJ; Silberstein, LE; Heslop, H; Weitz J; Anastasi, J. (2012). Hematology: Basic Principles and Practice (6th ed.). Elsevier. ISBN 978-1-4377-2928-3.
- Kawalec, Wanda (2015). Pediatria. Warsaw: PZWL. p. 1070. ISBN 978-83-200-4631-1.
- Elastase, neutrophil-expressed; ELANE. Online Mendelian Inheritance in Man. Johns Hopkins University.
- WAS gene; WAS. Online Mendelian Inheritance in Man. Johns Hopkins University.
- Zeidler C, Welte K (2002). "Kostmann syndrome and severe congenital neutropenia". Semin. Hematol. 39 (2): 82–8. doi:10.1053/shem.2002.31913. PMID 11957189.
- Xia J, Bolyard AA, Rodger E et al. Prevalence of mutations in ELANE, GFI1, HAX1, SBDS, WAS and G6PC3 in patients with severe congenital neutropenia. Br J Haematol. 2009;147(4):535. PMID|19775295
- Germeshausen, M., Deerberg, S., Peter, Y., et al. The spectrum of ELANE mutations and their implications in severe congenital and cyclic neutropenia. Hum. Mutat. 34: 905-914, 2013. PMID|23463630]
- Neutropenia, Severe Congenital, 2, Autosomal Dominant; SCN2. Online Mendelian Inheritance in Man. Johns Hopkins University.
- Neutropenia, Severe Congenital, 4, Autosomal Recessive; SCN4. Online Mendelian Inheritance in Man. Johns Hopkins University.
- Neutropenia, Severe Congenital, 5, Autosomal Recessive; SCN5. Online Mendelian Inheritance in Man. Johns Hopkins University.
- Stepensky P, Saada A, Cowan M, et al. (June 2013). "The Thr224Asn mutation in the VPS45 gene is associated with the congenital neutropenia and primary myelofibrosis of infancy". Blood. 121 (25): 5078–87. doi:10.1182/blood-2012-12-475566. PMID 23599270.
- Neutropenia, Severe Congenital, X-linked; SCNX. Online Mendelian Inheritance in Man. Johns Hopkins University.
- Online Mendelian Inheritance in Man (OMIM): Neutropenia, Severe congenital, 3, Autosomal recessive; SCN3 - 610738
- Christensen RD, Calhoun DA (2004). "Congenital neutropenia". Clin Perinatol. 31 (1): 29–38. doi:10.1016/j.clp.2004.03.011. PMID 15183654.
- Kostmann R (1956). "Infantile genetic agranulocytosis; agranulocytosis infantilis hereditaria". Acta Paediatr. 45 (Suppl 105): 1–78. doi:10.1111/j.1651-2227.1956.tb06875.x. PMID 13326376. S2CID 71420679.
- Klein, C.; Grudzien, M.; Appaswamy, G.; Germeshausen, M.; Sandrock, I.; Schäffer, A. A.; Rathinam, C.; Boztug, K.; Schwinzer, B.; Rezaei, N.; Bohn, G.; Melin, M.; Carlsson, G. R.; Fadeel, B.; Dahl, N.; Palmblad, J.; Henter, J. I.; Zeidler, C.; Grimbacher, B.; Welte, K. (Jan 2006). "HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease)". Nature Genetics. 39 (1): 86–92. doi:10.1038/ng1940. PMID 17187068. S2CID 22757727.
- McDermott DH, De Ravin SS, Jun HS et al. Severe congenital neutropenia resulting from G6PC3 deficiency with increased neutrophil CXCR4 expression and myelokathexis. Blood. 2010;116(15):2793. PMID|20616219
- Mir MA, Kochuparambil ST, Abraham RS, Rodriguez V, Howard M, Hsu AP, Jackson AE, Holland SM, Patnaik MM (April 2015). "Spectrum of myeloid neoplasms and immune deficiency associated with germline GATA2 mutations". Cancer Medicine. 4 (4): 490–9. doi:10.1002/cam4.384. PMC 4402062. PMID 25619630.
- Crispino JD, Horwitz MS (April 2017). "GATA factor mutations in hematologic disease". Blood. 129 (15): 2103–2110. doi:10.1182/blood-2016-09-687889. PMC 5391620. PMID 28179280.
- Hirabayashi S, Wlodarski MW, Kozyra E, Niemeyer CM (August 2017). "Heterogeneity of GATA2-related myeloid neoplasms". International Journal of Hematology. 106 (2): 175–182. doi:10.1007/s12185-017-2285-2. PMID 28643018.
- Newburger, Peter E.; Dale, David C. (July 2013). "Evaluation and Management of Patients With Isolated Neutropenia". Seminars in Hematology. 50 (3): 198–206. doi:10.1053/j.seminhematol.2013.06.010. PMC 3748385.
- Hoffbrand AV, Moss PA, Pettit JE (2005). Essential Haematology. Blackwell Publishing. ISBN 978-1-4051-3649-5.
- Klein, Christoph (2011). "Genetic Defects in Severe Congenital Neutropenia: Emerging Insights into Life and Death of Human Neutrophil Granulocytes". Annual Review of Immunology. 29: 399–413. doi:10.1146/annurev-immunol-030409-101259. PMID 21219176.
Further reading
- Deotare UR, Patel PD, Parikh RP, Bhagat EA (January 2012). "Uncommon cause of recurrent infections". Indian J Med Paediatr Oncol. 33 (1): 51–3. doi:10.4103/0971-5851.96971. PMC 3385280. PMID 22754210.