Griscelli syndrome
Griscelli syndrome is a rare autosomal recessive[1] disorder characterized by albinism (hypopigmentation) with immunodeficiency, that usually causes death by early childhood. Researchers have developed three different classifications of the form of disorder, characterised by different signs and symptoms. Type 1 Griscelli Syndrome is assosciated with severe brain function issues along with distinctive discolouring of the hair and skin. Type 2 Griscelli Syndrome have immune system abnormalities in addition to hypopigmentation of skin and hair. Finally, Type 3 is seen as those only affected by hypopigmentation of the skin and hair. This type is not associated with immune deficiencies or neurological abnormalities.
| Griscelli syndrome | |
|---|---|
| Other names | Partial albinism-immunodeficiency syndrome, Griscelli-Pruniéras syndrome, Chédiak-Higashi-like syndrome |
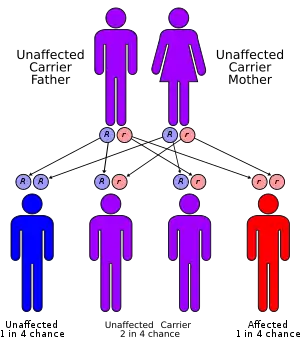 | |
| Griscelli syndrome has an autosomal recessive pattern of inheritance. | |
Signs and symptoms
Griscelli syndrome is defined by the characteristic hypopigmentation, with frequent pyogenic infection, enlargement of the liver and spleen, a low blood neutrophil level, low blood platelet level, and immunodeficiency. Very often there is also impaired natural killer cell activity, absent delayed-type hypersensitivity and a poor cell proliferation response to antigenic challenge. This may be caused by the loss of three different genes, each of which has different additional effects, resulting in three types of syndrome. Its inheritance is autosomal recessive.
Examination of the hair in this syndrome may be useful. Under light microscopy, these hairs exhibit bigger and irregular melanin granules, distributed mainly near the medulla. Under polarized light microscopy, the hairs appear monotonously white.[2]
Pathophysiology
In melanocytes, melanosomes (vesicles containing the pigment melanin) are transported on microtubules. They are then bound by Rab27A which recruits Slac2-a and myosin Va. This complex then transfers the melanosomes from the microtubules to actin filaments. This transfer is necessary for the transport of melanosomes from the perinuclear area to the cell periphery. The loss of any one of these proteins interrupts melanosome transport and results in the hypopigmentation.
However, these three proteins do not work together in other cells and RAB27A effectors may be 'mix and match.' For example, the knockout of Rab27 causes the hypopigmentation but also immunodeficiency due to deficiencies in cytotoxic killing activity in cytotoxic T cells (something that also depends on vesicle transport). While, the knockout of myosin Va does not cause immunodeficiency, but it does cause neural defects. Though some neural problems (i.e. brain damage) can be seen in Rab27A deficient children, this is thought to be a secondary effect of the immune problems, and not directly due to the lack of Rab27A. Munc13-4 has also drawn attention based on its involvement in causing bleeding manifestations in Griscelli syndrome.Munc13-4 through its interactions with Rab27a appears to be important for the dense granule release from platelets. The mutated Rab27a interaction with Munc13-4 is the cause of bleeding in type 2 Griscelli Syndrome.
Diagnosis
Types
Griscelli syndrome is a disorder of melanosome transport, and divided into several types:[3]: 866
| OMIM | Name | Gene |
|---|---|---|
| 214450 | Griscelli syndrome type 1 (Elejalde syndrome) | MYO5A |
| 607624 | Griscelli syndrome type 2 (Partial albinism with immunodeficiency) | RAB27A |
| 609227 | Griscelli syndrome type 3 | MLPH |
Management
Management and treatment is depended upon the type of syndrome one is diagnosed as having. Prognosis for long-term survival is however, relatively poor. Type 2 is usually rapidly fatal within 1 to 4 years without immediate, accelerated treatment.[4] Chemotherapy have achieved remissions however is sometimes ineffective for the treatment of the primary disease and can fail to control relapses. Allogenic bone-marrow transplantation (BMT) is the only known curative treatment in this disease. The severe neurological impairment and retarded development of the human does not improve with time.[5] During the accelerated phase, immunosuppressives can be used to control signs and symptoms. Since its discovery in 1976, only 40 citations have been found in modern literature.[6] In order to treat patients, the aggressive therapy strategy approach must always be taken for acute bacterial infections and prophylactic antibiotics. This assists in minimising possible effects and prolonging life expectancy.[7]
Eponym
It is named after Claude Griscelli, professor of pediatrics at Hôpital Necker Enfants-Malades in Paris (France).[8][5]
See also
References
- Griscelli C, Durandy A, Guy-Grand D, Daguillard F, Herzog C, Prunieras M (1978). "A syndrome associating partial albinism and immunodeficiency". Am. J. Med. 65 (4): 691–702. doi:10.1016/0002-9343(78)90858-6. PMID 707528.
- Valente NY, Machado MC, Boggio P, Alves AC, Bergonse FN, Casella E, Vasconcelos DM, Grumach AS, de Oliveira ZN (2006) Polarized light microscopy of hair shafts aids in the differential diagnosis of Chédiak-Higashi and Griscelli-Prunieras syndromes. Clinics (Sao Paulo) 61(4):327-332.
- James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 0-7216-2921-0.
- de Saint-Basile, Genevieve (November 2001). "Griscelli Syndrome" (PDF). Orphanet.
- Griscelli C, Prunieras M (1978). "Pigment dilution and immunodeficiency: a new syndrome". Int. J. Dermatol. 17 (10): 788–91. doi:10.1111/j.1365-4362.1978.tb05980.x. PMID 730432. S2CID 34244278.
- Rath, Sanjeev; Jain, Vivek; Marwaha, R. K.; Trehan, Amita; Rajesh, L. S.; Kumar, Vijay (February 2004). "Griscelli syndrome". The Indian Journal of Pediatrics. 71 (2): 173–175. doi:10.1007/bf02723104. ISSN 0019-5456. PMID 15053385. S2CID 33506748.
- "Immunodeficiency Search". immunodeficiency. Retrieved 2018-09-04.
- synd/3872 at Who Named It?
External links
- Griscelli syndrome type 1 at NIH's Office of Rare Diseases OMIM: 214450
- Griscelli syndrome type 2 at NIH's Office of Rare Diseases OMIM: 607624
- Griscelli syndrome type 3 at NIH's Office of Rare Diseases OMIM: 609227