Waardenburg syndrome
Waardenburg syndrome is a group of rare genetic conditions characterised by at least some degree of congenital hearing loss and pigmentation deficiencies, which can include bright blue eyes (or one blue eye and one brown eye), a white forelock or patches of light skin. These basic features constitute type 2 of the condition; in type 1, there is also a wider gap between the inner corners of the eyes called telecanthus, or dystopia canthorum.[1] In type 3, which is rare, the arms and hands are also malformed, with permanent finger contractures or fused fingers, while in type 4, the person also has Hirschsprung's disease.[2][3] There also exist at least two types (2E and PCWH) that can result in central nervous system (CNS) symptoms such as developmental delay and muscle tone abnormalities.[4]
| Waardenburg syndrome | |
|---|---|
| Other names | Klein–Waardenburg syndrome (type 3), Shah–Waardenburg syndrome (type 4) |
.jpg.webp) | |
| Facial features of Waardenburg syndrome type 1 (from Jan van der Hoeve's description, 1916) | |
| Specialty | Medical genetics |
The syndrome is caused by mutations in any of several genes that affect the division and migration of neural crest cells during embryonic development (though some of the genes involved also affect the neural tube).[5] Neural crest cells are stem cells left over after the closing of the neural tube that go on to form diverse non-CNS cells in different parts of the body, including melanocytes, various bones and cartilage of the face and inner ear and the peripheral nerves of the intestines.[6] Type 1 is caused by a mutation in the PAX3 gene, while the gene that most often causes type 2 when mutated is MITF.[1][7] Type 3 is a more severe presentation of type 1 and is caused by a mutation in the same gene, while type 4 is most often caused by a mutation in SOX10.[2][8] Mutations in other genes can also cause the different types, and some of these have been given their own lettered subtypes. Most types are autosomal dominant.
The estimated prevalence of Waardenburg syndrome is 1 in 42,000.[5][8] Types 1 and 2 are the most common, comprising approximately half and a third of cases, respectively, while type 4 comprises a fifth and type 3 less than 2% of cases.[8] An estimated 2–5% of congenitally deaf people have Waardenburg syndrome.[8] Descriptions of the syndrome date back to at least the first half of the 20th century, however it is named after Dutch ophthalmologist and geneticist Petrus Johannes Waardenburg, who described it in 1951.[9][10] Its subtypes were progressively discovered in the following decades and had genes attributed to them mostly in the 1990s and 2000s.
Signs and symptoms
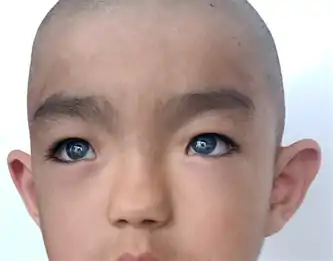
Waardenburg syndrome has multiple different types with some variations in symptoms, and symptoms can vary among those with the same type. The two features consistent across all types of Waardenburg syndrome are some degree of congenital sensorineural hearing loss and some degree of pigmentation deficiencies, most consistently in the eyes.[11]
Type 1
Type 1 is characterised by congenital sensorineural hearing loss, pigmentary deficiencies of the hair such as a white lock of hair (poliosis) in the front-centre of the head or premature greying, pigmentary deficiencies of the eyes such as different-coloured eyes (complete heterochromia iridum), multiple colours in an eye (sectoral heterochromia iridum) or brilliant blue eyes, patches of skin depigmentation, and a wider gap between the inner corners of the eyes called telecanthus or dystopia canthorum. Other facial features associated with type 1 can include a high nasal bridge, a flat nose tip, a unibrow (synophrys), smaller edges of the nostrils (alae) or a smooth philtrum.[1]
Type 2
The difference that defines type 2 from type 1 is that patients do not have the wider gap between the inner corners of the eyes (telecanthus/dystopia canthorum). Sensorineural hearing loss tends to be more common and more severe in this type.[7][12] By far the most common gene to cause this type when mutated is MITF (classified as type 2A). If two individuals with a mutation in this gene have a child carrying both mutations (homozygous), for which there is 25% chance, additional symptoms are present in the child, such as a hole in the iris (coloboma), small eyes (microphthalmia), hardened bones (osteopetrosis), macrocephaly, albinism and deafness.[7]
There have been two known patients identified with mutations in both copies of SNAI2 (classified as type 2D); these individuals presented with Waardenburg syndrome type 2 but did not have hair pigmentation deficiencies.[13]
When Waardenburg syndrome type 2 is caused by a mutation in SOX10 (classified as type 2E), it can on some occasions present with multiple neurological symptoms. These can include developmental delay, early childhood nystagmus, increased muscle tone, white matter anomalies or hypomyelination in the brain, autistic-like behaviour and the underdevelopment or complete absence of many inner-ear structures such as the vestibular system or cochlea. Lack of a sense of smell (anosmia) due to a missing olfactory bulb in the brain may also be present.[4]
Type 3
Also known as Klein–Waardenburg syndrome, or Waardenburg–Klein syndrome, type 3 has the same symptoms as type 1 (and is caused by mutations in the same gene) but has additional symptoms that affect the arms and hands. These can include joint contractures of the fingers (camptodactyly), due to underdeveloped muscles, as well as fused digits (syndactyly) or winged scapulae. Microcephaly and developmental delay are also possible.[2]
Type 4
Also known as Shah–Waardenburg syndrome, or Waardenburg–Shah syndrome, type 4 has most of the same features as type 2 (i.e. no telecanthus, or apparent wider eye gap), but with the addition of Hirschsprung's disease, which is a congenital lack of nerves in the intestines leading to bowel dysfunction.[3][14] Additionally, hearing loss is not as common as in type 2.[3] Rarely, cleft lip has been reported in this form of Waardenburg syndrome.[15]
Type 4 can also be caused by a mutation in SOX10 (the same gene as in type 2E), in which it is known as type 4C; hearing loss is very common and severe in this type.[16]
PCWH
A mutation in SOX10, the gene involved in type 2E and type 4C, can sometimes result in the symptoms of both types (neurological symptoms, as sometimes seen in type 2E, and Hirschsprung's disease, as seen in type 4). When this happens, it is called peripheral demyelinating neuropathy–central dysmyelinating leukodystrophy–Waardenburg syndrome–Hirschsprung disease (PCWH).[17][18]
Cause
.svg.png.webp)
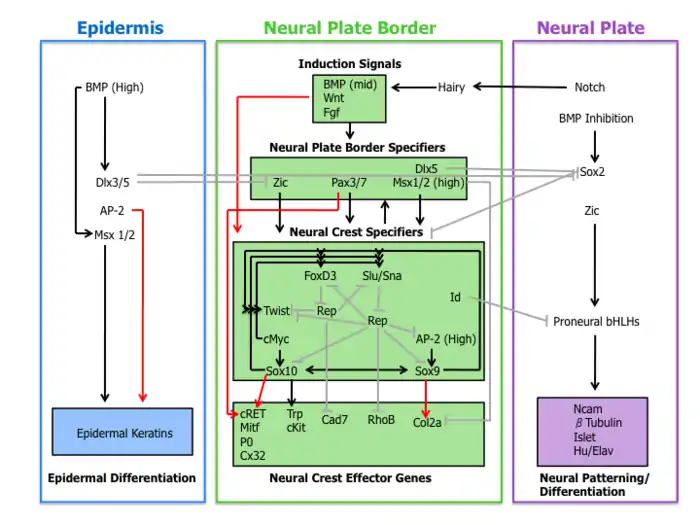
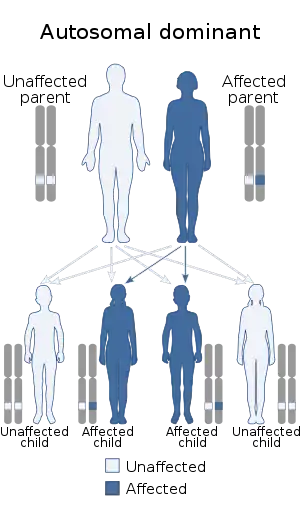

Waardenburg syndrome is caused by mutations in any of several genes that affect the operation of neural crest cells in embryonic development. Most types of Waardenburg syndrome are caused by autosomal dominant mutations. The few that are autosomal recessive are rare. In most cases, an affected person has inherited it from one parent with one of the dominant forms of the condition. A small percentage of cases result from spontaneous new mutations in the gene, where no family history of the condition exists.
The neural crest is a group of temporary migratory cells that are left over after the neural tube has closed (neurulation), around the fourth week of embryonic development. They are responsible for differentiating into a diverse group of cells that reach different areas of the body. The neural tube and neural crest are derived from the ectoderm; the neural tube goes on to form the brain and spinal cord, while the neural crest cells eventually go on to form various bones and cartilage of the skull and face by migrating through the pharyngeal arches. They also differentiate into the stria vascularis of the cochlea, the nerves and glia of the intestines (myenteric plexus), Schwann cells, which myelinate the peripheral nervous system to allow sufficient conductivity, odontoblasts, which produce dentin deep in the teeth, some neuroendocrine cells, connective tissue around the salivary, lacrimal, pituitary, thymus and thyroid glands, connective tissue of the eye, such as the stroma of the iris and cornea and the trabecular meshwork,[19] and melanocytes, including those in the stroma of the iris that give rise to brown eye colour through melanin. Neural crest cells also have a role in muscle formation, including the wall muscle of certain cardiac arteries.[6]
Causes of subtypes
- Type 1 is caused by an autosomal dominant mutation in the gene PAX3.[1] PAX3, or paired box 3, is a transcription factor that has a role in maintaining an open window of time for certain neural crest cells (such as those of the head and eyes) to divide and migrate before their terminal differentiation (i.e. to maintain them in the stem-cell state). Mutations in this gene therefore prematurely arrest their division and migration, resulting in a minor lack of development of certain face cartilage and bones, as well as underdeveloped inner-ear structures and a lack of melanocytes in the iris stroma. Some evidence shows that PAX3 also regulates cells from before the neural crest forms, i.e. the neural tube, since mice with loss-of-function mutations in one of the copies of PAX3 have neural tube defects such as spina bifida or exencephaly.[5]
- Type 2 is caused by a mutation in any of a range of genes, the most common being MITF, when it is classed as type 2A.
- Type 2A is caused by an autosomal dominant mutation in the gene MITF.[7] MITF, or microphthalmia-associated transcription factor, has a more specialised role in the neural crest and is more strictly involved after the neural crest forms (PAX3 and SOX10 have been found to activate MITF).[20] It is known to allow melanocytes, osteoclasts, mast cells and retinal pigment epithelial cells to divide and migrate. The involvement in osteoclasts explains why mutations in both copies of MITF can lead to bone hardening (osteopetrosis), as the osteoclasts are responsible for breaking down bone. MITF also activates transcription of tyrosinase, the enzyme that performs the first step in the creation of melanin (oxidising tyrosine). A mutation in a copy of MITF can also lead to Tietz syndrome, which is distinguished from Waardenburg syndrome by uniform albinism instead of patchy depigmentation.[5]
- Type 2B is caused by an autosomal dominant mutation in an unknown gene on chromosome 1 in the locus range of 1p21–1p13.3. The gene has been provisionally termed WS2B.[21][22]
- Type 2C is caused by an autosomal dominant mutation in an unknown gene on chromosome 8 in the locus of 8p23. The gene has been provisionally termed WS2C.[23][24]
- Type 2D is caused by an autosomal recessive mutation in both copies of the gene SNAI2. The study that discovered this association found that SNAI2 is activated by MITF as part of neural crest development, and this explained why mutations in MITF cause Waardenburg syndrome, as it results in a lack of activation of SNAI2. Mutations in a single copy of SNAI2 have also been found to cause patches of hair depigmentation (piebaldism) without any other symptoms.[25]
- Type 2E is caused by an autosomal dominant mutation in the gene SOX10.[4]
- Rarely, a mutation in a gene other than those currently known may be responsible for a Waardenburg syndrome with features of type 2. This is usually initially classified as simply type 2 but may be given its own subtype once a gene or locus is identified and established.[7]
- Type 3 is caused by a mutation in the gene PAX3, the same gene as in type 1.[2] It can be inherited in an autosomal dominant or autosomal recessive manner; it is possible for two parents with Waardenburg syndrome type 1 to have a child carrying both mutated copies of the PAX3 gene (25% chance) and present with Waardenburg syndrome type 3. A missense mutation has been documented to have this effect. However, it is also possible for Waardenburg syndrome type 3 to present spontaneously with just one mutated copy of PAX3. A deletion of the paired domain region of the gene has been documented to have this effect.[26][5] However, no major correlation has been found between type of mutation and disease severity. Severity tends to be dictated by mutations in other genes (epistasis), as evidenced by distinct familial patterns in severity not tied to Waardenburg mutation type.[5] Mutations in both copies of PAX3 have sometimes led to death before or shortly after birth, and mice with loss-of-function mutations in both copies of the gene do not survive.[5]
- Type 4 is caused by a mutation in any of a range of genes, the most common being SOX10, when it is classed as type 4C.
- Type 4A is caused by an autosomal dominant or autosomal-recessive mutation in the gene EDNRB.[15]
- Type 4B is caused by an autosomal dominant or autosomal-recessive mutation in the gene EDN3.[27]
- Type 4C is caused by an autosomal dominant or autosomal-recessive mutation in the gene SOX10, the same gene as in type 2E.[16]
A study was done on a rare case of a double heterozygous child with each parent having only single mutations in MITF or PAX3. The effect of double heterozygous mutations in the genes MITF and PAX3 in WS1 and WS2 can increase the pigment-affected symptoms. It leads to the conclusion that the double mutation of MITF is associated with the extremity of Waardenburg syndrome and may affect the phenotypes or symptoms of the syndrome.[28]
Classification table
| Type | OMIM | Gene | Locus | Inheritance |
|---|---|---|---|---|
| Type 1 (WS1) | 193500 | PAX3 | 2q36.1[29] | Autosomal dominant |
| Type 2A (WS2A, originally WS2) | 193510 | MITF | 3p14.1–p12.3 | Autosomal dominant |
| Type 2B (WS2B) | 600193 | WS2B | 1p21–p13.3 | Autosomal dominant |
| Type 2C (WS2C) | 606662 | WS2C | 8p23 | Autosomal dominant |
| Type 2D (WS2D) | 608890 | SNAI2 | 8q11 | Autosomal recessive |
| Type 2E (WS2E) | 611584 | SOX10 | 22q13.1 | Autosomal dominant |
| Type 3 (WS3) | 148820 | PAX3 | 2q36.1 | Autosomal dominant or autosomal recessive |
| Type 4A (WS4A) | 277580 | EDNRB | 13q22 | Autosomal dominant or autosomal recessive |
| Type 4B (WS4B) | 613265 | EDN3 | 20q13 | Autosomal dominant or autosomal recessive |
| Type 4C (WS4C) | 613266 | SOX10 | 22q13.1 | Autosomal dominant |
Treatment
There is currently no treatment or cure for Waardenburg syndrome. The symptom most likely to be of practical importance is deafness, and this is treated as any other irreversible deafness would be. In marked cases, there may be cosmetic issues. Other abnormalities (neurological, structural, Hirschsprung's disease) associated with the syndrome are treated symptomatically.
Epidemiology
The prevalence of all types of Waardenburg syndrome is estimated at around 1 in 42,000.[5][8] Types 1 and 2 are by far the most common, with type 1 appearing to be slightly more common.[30][31] In a 2015 review looking at 417 patients, type 1 was found to be the most common type, encompassing around half of all cases (47%), while type 2 was the second-most common type, encompassing around a third (33%).[8] The vast majority (around 85%) of type 2 cases are type 2A.[8] The prevalence of type 2B is unknown, as it was only reported in one 1996 study.[22] Type 2C has so far only been found in one Italian family,[23][24] and type 2D had only been found in 2 unrelated patients as of 2018.[13][8][32] The number of known cases of type 2E that involved neurological abnormalities was reported to be 23 as of 2017,[33] while the number of the rest is unknown. Type 3 is rarer than types 1, 2 and 4,[34] comprising less than 2% of cases.[8] Type 4 appears to encompass around a fifth of cases (19%). Of its subtypes, type 4C is by far the most common (about 71% of type 4), followed by type 4A (19%) and type 4B (10%).[8]
It is estimated that Waardenburg syndrome is present in 2–5% of congenitally deaf people. Congenital deafness comprises around half of deafness as a whole.[8] About 1 in 30 students in schools for the deaf have Waardenburg syndrome. The variable presentation of the syndrome makes it difficult to arrive at precise figures for its prevalence.[8]
History
Early descriptions
In 1916, Dutch ophthalmologist Jan van der Hoeve (1878–1952) described a pair of twin girls with deafness and a particular type of blepharophimosis, believed to be the dystopia canthorum found in Waardenburg syndrome types 1 and 3.[8][35] Blepharophimosis describes eyelids which are underdeveloped such that they permanently cover part of the eyes.
In 1926, German physician Irmgard Mende described a family of four generations in which five children had symptoms of depigmentation of hair, skin and eyes, deafness and a "mongoloid" appearance. (Waardenburg later attributed this description to the dystopia canthorum.)[36][35] This later led to the synonym Mende syndrome being recorded in some databases.[11][37]
In 1929, Dutch physician K. T. A. Halbertsma described a familial pattern to dystopia canthorum,[38][35] and in 1930, Italian physician Vincenzo Gualdi[39] (1891–1976) also confirmed a hereditary pattern to dystopia canthorum.[36] This later led to the synonym Van der Hoeve–Halbertsma–Waardenburg–Gualdi syndrome being recorded in some databases.[11]
In 1947, Swiss ophthamologist David Klein (1908–1993) first reported a patient with bilateral deafness, pigmentation deficiencies, characteristic facial features and malformation of the arms. Although this was the first full description of a patient with Waardenburg syndrome type 3, contemporary clinicians did not consider the syndrome he described to be the same as that described by Waardenburg four years later, in part due to how severe the arm malformations were in his patient.[40]
The syndrome was first fully formalised and described by Dutch ophthalmologist and geneticist Petrus Johannes Waardenburg (1886–1979) in 1951.[9][10] The condition he described is now categorised as Waardenburg syndrome type 1.
Descriptions of subtypes
Type 2 was first established in 1971 when a study noticed that some Waardenburg syndrome patients did not have dystopia canthorum.[7][41] A 1977 study confirmed a familial pattern to this other presentation.[7] Two 1994 studies first confirmed a link between this type of Waardenburg syndrome and mutations in the MITF gene (now classed as type 2A), located on chromosome 3 at locus 3p14.1–p12.3.[7]
Type 2B was first established in 1994 when the same study which found mutations in MITF in patients with Waardenburg syndrome type 2 also found that some patients did not have any mutations in this region.[21][42] A second 1994 study found a link to chromosome 1 in the locus 1p21–p13.3. This became known as type 2B of the condition (with the gene designated WS2B), however it has not been documented since, and the gene responsible remains unknown.[21][22]
Type 2C was established in 2001 when a study of an Italian family with Waardenburg syndrome type 2 features found that they were due to an unknown gene on chromosome 8 at locus 8q23 that had been broken by a chromosomal translocation. The study established a provisional name for the gene, WS2C. However, mutations in this region in Waardenburg syndrome patients have not been found since.[23][24]
Type 2D was established in 2002 when a study looking to find mutations in the human version of the SNAI2 gene, known to cause depigmentation in mice, found deletions of both copies of this gene in two unrelated individuals with Waardenburg syndrome type 2. Mutations in both copies of this gene have not been found in those with Waardenburg syndrome type 2 since.[8]
Type 2E was first established in 1996 when a study identified a girl with symptoms of Waardenburg syndrome type 2 but with additional underdevelopment of the front of the eye, leading to blindness. In 1999, it was found that she had a mutation in her SOX10 gene, and later studies confirmed the association between mutations in this gene and this phenotype, as well as neurological symptoms such as developmental delay.[4]
Type 3 was first given its name by Goodman et al. in 1981, in collaboration with Klein, in which they established the association with arm abnormalities first reported by Klein in 1947.[40] Mutations in PAX3 were first linked to this phenotype in 1992.[2]
The comorbidity with Hirschsprung's disease, which would later constitute type 4, was first noticed in various studies in the 1970s. Indian paediatrician Krishnakumar Shah and his associates first outlined the syndrome as a possible variant of Waardenburg syndrome in 1981.[43] The variant was first attributed to a mutation in EDNRB in 1994 (now classed as type 4A).[3] Type 4B was established in 1996 when mutations in EDN3 were found to lead to this type of Waardenburg syndrome,[27] and type 4C was first established in 1998 when mutations in SOX10 were also found to lead to this type.[16]
Society and culture
Popular culture
- The 2001 novel Shock by Robin Cook mentions a character with the disorder.[44]
- Enzo MacLeod, protagonist of Peter May's 2006–2017 book series The Enzo Files, has Waardenburg syndrome. His eyes are different colors, and he has a white streak in his hair.[45][46]
- In the 2011 season 6 episode of Bones "The Signs in the Silence", the team must solve a case in which the suspected killer has Waardenburg syndrome.[47]
- The 2013 book Reconstructing Amelia by Kimberly McCreight features several characters with Waardenburg symptoms.[48]
- The 2014 book Closer Than You Think by Karen Rose features three characters, siblings, with Waardenburg syndrome.[49]
- The 2017 book Murder at the Mayan Temple by M.J. Mandrake features several characters with Waardenburg syndrome.[50]
- The 2019 novel The Whisper Network by Chandler Baker uses the syndrome as a plot point.
Notable people
- Canadian YouTube vlogger Stef Sanjati has Waardenburg syndrome type 1.[51]
Other animals

Waardenburg syndrome type 2A (with a mutation in MITF) has been found in dogs, Fleckvieh cattle, minks, mice and a golden hamster.[52] Degeneration of the cochlea and saccule, as seen in Waardenburg syndrome, has also been found in deaf white cats, Dalmatians and other dog breeds, white minks and mice.[53]
Domesticated cats with blue eyes and white coats are often completely deaf.[54] Deafness is far more common in white cats than in those with other coat colors. According to the ASPCA Complete Guide to Cats, "17 to 20 percent of white cats with non-blue eyes are deaf; 40 percent of "odd-eyed" white cats with one blue eye are deaf; and 65 to 85 percent of blue-eyed white cats are deaf."[55] Although few studies have been done to link this to genes known to be involved in human Waardenburg syndrome, a genetic disruption to neural crest development would lead to this presentation in cats as well.[56] One of the genes that leads to deafness and a white coat in cats when mutated, KIT,[57] has been found to increase MITF expression.[58]
Lethal white syndrome is a syndrome in horses caused by mutations in both copies of EDNRB. It leads to death from intestinal pseudo-obstruction due to Hirschsprung's disease. A mutation in a single copy of EDNRB, however, as in Waardenburg syndrome type 4A, produces the patchy white overo coat with deafness.[59]
Ferrets with Waardenburg syndrome have a small white stripe along the top or back of the head and sometimes down the back of the neck (known as a "blaze" coat pattern), or a solid-white head from nose to shoulders (known as a "panda" coat pattern). Affected ferrets often have a very slightly flatter skull and wider-set eyes than healthy ferrets. As healthy ferrets have poor hearing, deafness may only be detected by lack of reaction to loud noises. As this is an inherited disorder, affected animals should not be used for breeding. A study of the correlation between coat variations and deafness in European ferrets found, "All (n=27) panda, American panda, and blaze ferrets were deaf."[60]
See also
- Chédiak–Higashi syndrome, a similar syndrome including immunodeficiency and peripheral neuropathy
- Tietz syndrome, a condition similar to Waardenburg syndrome type 2 involving uniform albinism (caused by mutations in MITF)
- Vogt–Koyanagi–Harada disease, an autoimmune disease causing uveitis, patchy depigmentation and inner ear symptoms
References
- "OMIM Entry - # 193500 - WAARDENBURG SYNDROME, TYPE 1; WS1". omim.org. Retrieved 2019-12-07.
- "OMIM Entry - # 148820 - WAARDENBURG SYNDROME, TYPE 3; WS3". omim.org. Retrieved 2019-12-07.
- "OMIM Entry - # 277580 - WAARDENBURG SYNDROME, TYPE 4A; WS4A". omim.org. Retrieved 2019-12-07.
- "OMIM Entry - # 611584 - WAARDENBURG SYNDROME, TYPE 2E; WS2E". omim.org. Retrieved 2019-12-07.
- Pingault, Véronique; Ente, Dorothée; Moal, Florence Dastot-Le; Goossens, Michel; Marlin, Sandrine; Bondurand, Nadège (2010). "Review and update of mutations causing Waardenburg syndrome". Human Mutation. 31 (4): 391–406. doi:10.1002/humu.21211. ISSN 1098-1004. PMID 20127975. S2CID 12278025.
- "Neural Crest Development - Embryology". embryology.med.unsw.edu.au. Retrieved 2019-12-13.
- "OMIM Entry - # 193510 - WAARDENBURG SYNDROME, TYPE 2A; WS2A". omim.org. Retrieved 2019-12-07.
- Song, J.; Feng, Y.; Acke, F. R.; Coucke, P.; Vleminckx, K.; Dhooge, I. J. (2016). "Hearing loss in Waardenburg syndrome: a systematic review". Clinical Genetics. 89 (4): 416–425. doi:10.1111/cge.12631. ISSN 1399-0004. PMID 26100139. S2CID 23834634.
- Chandra Mohan, Setty. L. N. (2018-09-01). "Case of Waardenburg Shah syndrome in a family with review of literature". Journal of Otology. 13 (3): 105–110. doi:10.1016/j.joto.2018.05.005. ISSN 1672-2930. PMC 6291636. PMID 30559775.
- Waardenburg PJ (September 1951). "A New Syndrome Combining Developmental Anomalies of the Eyelids, Eyebrows and Noseroot with Pigmentary Anomalies of the Iris and Head Hair and with Congenital Deafness; Dystopia canthi medialis et punctorum lacrimalium lateroversa, Hyperplasia supercilii medialis et radicis nasi, Heterochromia iridum totalis sive partialis, Albinismus circumscriptus (leucismus, poliosis) et Surditas congenita (surdimutitas)". Am. J. Hum. Genet. 3 (3): 195–253. PMC 1716407. PMID 14902764.
- "Waardenburg syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2018-04-17.
- "Waardenburg syndrome". Genetics Home Reference. October 2012.
- "OMIM Entry - # 608890 - WAARDENBURG SYNDROME, TYPE 2D; WS2D". omim.org. Retrieved 2019-12-07.
- Baral V, Chaoui A, Watanabe Y, Goossens M, Attie-Bitach T, Marlin S, Pingault V, Bondurand N (2012). "Screening of MITF and SOX10 regulatory regions in Waardenburg syndrome type 2". PLOS ONE. 7 (7): e41927. Bibcode:2012PLoSO...741927B. doi:10.1371/journal.pone.0041927. PMC 3407046. PMID 22848661.
- Pierpont, J. W.; St Jacques, D.; Seaver, L. H.; Erickson, R. P. (March 1995). "A family with unusual Waardenburg syndrome type I (WSI), cleft lip (palate), and Hirschsprung disease is not linked to PAX 3". Clinical Genetics. 47 (3): 139–143. doi:10.1111/j.1399-0004.1995.tb03946.x. ISSN 0009-9163. PMID 7634536. S2CID 24530898.
- "OMIM Entry - # 613266 - WAARDENBURG SYNDROME, TYPE 4C; WS4C". omim.org. Retrieved 2019-12-07.
- "Orphanet: Search a disease". www.orpha.net. Retrieved 2019-12-10.
- Verheij, Johanna B. G. M.; Sival, Deborah A.; van der Hoeven, Johannes H.; Vos, Yvonne J.; Meiners, Linda C.; Brouwer, Oebele F.; van Essen, Anthonie J. (January 2006). "Shah-Waardenburg syndrome and PCWH associated with SOX10 mutations: a case report and review of the literature". European Journal of Paediatric Neurology. 10 (1): 11–17. doi:10.1016/j.ejpn.2005.10.004. ISSN 1090-3798. PMID 16504559.
- Williams, Antionette L.; Bohnsack, Brenda L. (June 2015). "Neural Crest Derivatives in Ocular Development: Discerning the Eye of the Storm". Birth Defects Research Part C: Embryo Today: Reviews. 105 (2): 87–95. doi:10.1002/bdrc.21095. ISSN 1542-975X. PMC 5262495. PMID 26043871.
- Kawakami, Akinori; Fisher, David E. (June 2017). "The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology". Laboratory Investigation; A Journal of Technical Methods and Pathology. 97 (6): 649–656. doi:10.1038/labinvest.2017.9. ISSN 1530-0307. PMID 28263292.
- "OMIM Entry - % 600193 - WAARDENBURG SYNDROME, TYPE 2B; WS2B". www.omim.org. Retrieved 2019-12-23.
- Lalwani, A. K.; San Agustin, T. B.; Wilcox, E. R. (1994-09-01). "A locus for Waardenburg syndrome type II maps to chromosome 1p13.3-2.1". American Journal of Human Genetics. 55 (Suppl.3). OSTI 133315.
- "OMIM Entry - % 606662 - WAARDENBURG SYNDROME, TYPE 2C; WS2C". omim.org. Retrieved 2019-12-07.
- Selicorni, Angelo; Guerneri, Silvana; Ratti, Antonia; Pizzuti, Antonio (January 2002). "Cytogenetic mapping of a novel locus for type II Waardenburg syndrome". Human Genetics. 110 (1): 64–67. doi:10.1007/s00439-001-0643-9. ISSN 0340-6717. PMID 11810298. S2CID 24411957.
- Sánchez‐Martín, Manuel; Pérez‐Losada, Jesús; Rodríguez‐García, Arancha; González‐Sánchez, Belén; Korf, Bruce R.; Kuster, W.; Moss, Celia; Spritz, Richard A.; Sánchez‐García, I. (2003). "Deletion of the SLUG (SNAI2) gene results in human piebaldism". American Journal of Medical Genetics Part A. 122A (2): 125–132. doi:10.1002/ajmg.a.20345. ISSN 1552-4833. PMID 12955764. S2CID 33811699.
- Tekin, M.; Bodurtha, J. N.; Nance, W. E.; Pandya, A. (2001). "Waardenburg syndrome type 3 (Klein–Waardenburg syndrome) segregating with a heterozygous deletion in the paired box domain of PAX3: a simple variant or a true syndrome?". Clinical Genetics. 60 (4): 301–304. doi:10.1034/j.1399-0004.2001.600408.x. ISSN 1399-0004. PMID 11683776. S2CID 24025063.
- "OMIM Entry - # 613265 - WAARDENBURG SYNDROME, TYPE 4B; WS4B". omim.org. Retrieved 2019-12-07.
- Yang T, Li X, Huang Q, Li L, Chai Y, Sun L, Wang X, Zhu Y, Wang Z, Huang Z, Li Y, Wu H (January 2013). "Double heterozygous mutations of MITF and PAX3 result in Waardenburg syndrome with increased penetrance in pigmentary defects". Clin. Genet. 83 (1): 78–82. doi:10.1111/j.1399-0004.2012.01853.x. PMID 22320238. S2CID 34173541.
- Tsukamoto K, Nakamura Y, Niikawa N (March 1994). "Isolation of two isoforms of the PAX3 gene transcripts and their tissue-specific alternative expression in human adult tissues". Hum. Genet. 93 (3): 270–4. doi:10.1007/bf00212021. PMID 7545913. S2CID 36749688.
- "Orphanet: Waardenburg syndrome type 1". www.orpha.net. Retrieved 2019-12-10.
- "Waardenburg syndrome type II" (PDF). Orphanet. 2005. Retrieved 10 December 2019.
- Saleem, Mohammed D. (2019). "Biology of human melanocyte development, Piebaldism, and Waardenburg syndrome". Pediatric Dermatology. 36 (1): 72–84. doi:10.1111/pde.13713. ISSN 1525-1470. PMID 30561083.
- Bogdanova‐Mihaylova, Petya; Alexander, Michael D.; Murphy, Raymond P. J.; Murphy, Sinéad M. (2017). "Waardenburg syndrome: a rare cause of inherited neuropathy due to SOX10 mutation". Journal of the Peripheral Nervous System. 22 (3): 219–223. doi:10.1111/jns.12221. ISSN 1529-8027. PMID 28544110. S2CID 13676694.
- "Orphanet: Waardenburg syndrome type 3". www.orpha.net. Retrieved 2019-12-10.
- De Haas, E. B. H.; Tan, K. E. W. P. (1966-01-01). "Waardenburg's syndrome". Documenta Ophthalmologica. 21 (1): 239–282. doi:10.1007/BF00184136. ISSN 1573-2622. S2CID 10163905.
Waardenburg (1951, 1957, 1961) has expressed the belief that all these cases of uncomplicated blepharophimosis do in fact belong to his syndrome and that this type of dystopia canthorum does not occur as a separate trait. ... With regard to Mende's cases, [Waardenburg] believes that the 'mongoloid component' in these patients was in actuality due to dystopia canthorum.
- Klein, D. (February 1983). "Historical background and evidence for dominant inheritance of the Klein-Waardenburg syndrome (type III)". American Journal of Medical Genetics. 14 (2): 231–239. doi:10.1002/ajmg.1320140205. ISSN 0148-7299. PMID 6340503.
- Stedman, Thomas Lathrop (2005). Stedman's Medical Eponyms. Lippincott Williams & Wilkins. ISBN 978-0-7817-5443-9.
- "Over twee op elkaar gelijkende, in wezen echter verschillende aangeboren oogafwijkingen". Nederlands Tijdschrift voor Geneeskunde (in Dutch). 2009-12-02. Retrieved 2019-12-10.
- Annuario del Ministero dell'Educazione nazionale (in Italian). Provveditorato generale dello Stato. 1935. p. 142.
- Goodman, R. M.; Lewithal, I.; Solomon, A.; Klein, D. (April 1982). "Upper limb involvement in the Klein-Waardenburg syndrome". American Journal of Medical Genetics. 11 (4): 425–433. doi:10.1002/ajmg.1320110407. ISSN 0148-7299. PMID 7091186.
- Arias, S. (March 1971). "Genetic heterogeneity in the Waardenburg syndrome". Birth Defects Original Article Series. 07 (4): 87–101. ISSN 0547-6844. PMID 5006208.
- Hughes, A. E.; Newton, V. E.; Liu, X. Z.; Read, A. P. (August 1994). "A gene for Waardenburg syndrome type 2 maps close to the human homologue of the microphthalmia gene at chromosome 3p12-p14.1". Nature Genetics. 7 (4): 509–512. doi:10.1038/ng0894-509. ISSN 1061-4036. PMID 7951321. S2CID 2913481.
- Shah, Krishnakumar N.; Dalal, Subhash J.; Desai, Meena P.; Sheth, Prem N.; Joshi, Nana C.; Ambani, Lalit M. (1981-09-01). "White forelock, pigmentary disorder of irides, and long segment Hirschsprung disease: Possible variant of Waardenburg syndrome". The Journal of Pediatrics. 99 (3): 432–435. doi:10.1016/S0022-3476(81)80339-3. ISSN 0022-3476. PMID 7264803.
- Cook, Robin (2011-12-12). Shock. Pan Macmillan. p. 175. ISBN 978-1-4472-1796-1.
- May, Peter (2013-05-30). Extraordinary People: Enzo Macleod 1. Quercus. p. 32. ISBN 978-1-78206-885-3.
- May, Peter (2013-06-13). Blacklight Blue: Enzo Macleod 3. Quercus. p. 76. ISBN 978-1-78206-887-7.
- "Bones Recap 6.21 "The Signs in the Silence" – Persephone Magazine". Retrieved 2019-12-14.
- McCreight, Kimberly (2013-04-01). Reconstructing Amelia. Simon and Schuster. ISBN 978-1-4711-2944-5.
- Rose, Karen (2014-11-06). Closer Than You Think (The Cincinnati Series Book 1). Headline. ISBN 978-0-7553-8999-5.
- mynonie. "Meet a nice sleuth". Amazon.com. Retrieved 2019-12-14.
- Edwards, Lucy (2018). "'I get you're transgender, but what's up with your face?'". BBC News. Retrieved 2018-01-28.
- MARKAKIS, MARIOS N.; SOEDRING, VIBEKE E.; DANTZER, VIBEKE; CHRISTENSEN, KNUD; ANISTOROAEI, RAZVAN (2014-08-01). "Association of MITF gene with hearing and pigmentation phenotype in Hedlund white American mink (Neovison vison)". Journal of Genetics. 93 (2): 477–481. doi:10.1007/s12041-014-0370-3. hdl:10067/1211550151162165141. ISSN 0973-7731. PMID 25189243. S2CID 16725018.
- Strain, George M. (2015). "The Genetics of Deafness in Domestic Animals". Frontiers in Veterinary Science. 2: 29. doi:10.3389/fvets.2015.00029. ISSN 2297-1769. PMC 4672198. PMID 26664958.
- Webb AA, Cullen CL (June 2010). "Coat color and coat color pattern-related neurologic and neuro-ophthalmic diseases". Can. Vet. J. 51 (6): 653–7. PMC 2871368. PMID 20808581.
- Richards J (1999). ASPCA Complete Guide to Cats: Everything You Need to Know About Choosing and Caring for Your Pet. Chronicle Books. p. 71. ISBN 9780811819299.
- Omenn, Gilbert S.; McKusick, Victor A.; Gorlin, Robert J. (1979). "The association of Waardenburg syndrome and Hirschsprung megacolon". American Journal of Medical Genetics. 3 (3): 217–223. doi:10.1002/ajmg.1320030302. ISSN 1096-8628. PMID 484594.
The deaf, blue-eyed, white cat, noted by Bree [1829] and by Darwin [1892] and studied histologically at the turn of the century [Alexander, 1900], has a variable clinical and histologic picture, due to either of two autosomal dominant genes ... These pleiotropic effects of single genes may be explained by effects on the neural crest cells involved in the origin of all the tissues affected in Waardenburg syndrome [Weston, 1969].
- David, Victor A.; Menotti-Raymond, Marilyn; Wallace, Andrea Coots; Roelke, Melody; Kehler, James; Leighty, Robert; Eizirik, Eduardo; Hannah, Steven S.; Nelson, George; Schäffer, Alejandro A.; Connelly, Catherine J. (2014-10-01). "Endogenous Retrovirus Insertion in the KIT Oncogene Determines White and White spotting in Domestic Cats". G3: Genes, Genomes, Genetics. 4 (10): 1881–1891. doi:10.1534/g3.114.013425. ISSN 2160-1836. PMC 4199695. PMID 25085922.
- Lee, Youl-Nam; Brandal, Stephanie; Noel, Pierre; Wentzel, Erik; Mendell, Joshua T.; McDevitt, Michael A.; Kapur, Reuben; Carter, Melody; Metcalfe, Dean D.; Takemoto, Clifford M. (2011-03-31). "KIT signaling regulates MITF expression through miRNAs in normal and malignant mast cell proliferation". Blood. 117 (13): 3629–3640. doi:10.1182/blood-2010-07-293548. ISSN 0006-4971. PMC 3072881. PMID 21273305.
- Magdesian, K. Gary; Williams, D. Colette; Aleman, Monica; LeCouteur, Richard A.; Madigan, John E. (2009-11-15). "Evaluation of deafness in American Paint Horses by phenotype, brainstem auditory-evoked responses, and endothelin receptor B genotype". Journal of the American Veterinary Medical Association. 235 (10): 1204–1211. doi:10.2460/javma.235.10.1204. ISSN 0003-1488. PMID 19912043.
- Piazza S, Abitbol M, Gnirs K, Huynh M, Cauzinille L (May 2014). "Prevalence of deafness and association with coat variations in client-owned ferrets". J. Am. Vet. Med. Assoc. 244 (9): 1047–52. doi:10.2460/javma.244.9.1047. PMID 24739114.
External links
- GeneReviews/NCBI/NIH/UW entry on Waardenburg Syndrome Type I
- OMIM Genetic disorder catalog — Waardenburg syndrome