Methylmalonyl-CoA mutase deficiency
Methylmalonyl-CoA mutase is a mitochondrial homodimer apoenzyme (EC. 5. 4.99.2) that focuses on the catalysis of methylmalonyl CoA to succinyl CoA. The enzyme is bound to adenosylcobalamin, a hormonal derivative of vitamin B12 in order to function. Methylmalonyl-CoA mutase deficiency[2] is caused by genetic defect in the MUT [3] gene responsible for encoding the enzyme. Deficiency in this enzyme accounts for 60% of the cases of methylmalonic acidemia.[4]
| Methylmalonyl-CoA mutase deficiency | |
|---|---|
| Other names | MCM Deficiency [1] |
| Methylmalonyl-CoA mutase | |||||||
|---|---|---|---|---|---|---|---|
 Rendering based on PDB 2XIJ. | |||||||
| Identifiers | |||||||
| Symbol | MMUT | ||||||
| Alt. symbols | MCM, MUT | ||||||
| NCBI gene | 4594 | ||||||
| HGNC | 7526 | ||||||
| OMIM | 609058 | ||||||
| RefSeq | NP_000246 | ||||||
| UniProt | P22033 | ||||||
| Other data | |||||||
| EC number | 5.4.99.2 | ||||||
| Locus | Chr. 6 p21 | ||||||
| |||||||
| methylmalonyl-CoA mutase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| EC no. | 5.4.99.2 | ||||||||
| CAS no. | 9023-90-9 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
Symptoms
People with methylmalonyl CoA mutase deficiency exhibit many symptoms similar to other diseases involving inborn errors of metabolism.
Newborn babies experience with vomiting, acidosis, hyperammonemia, hepatomegaly (enlarged livers), hyperglycinemia (high glycine levels), and hypoglycemia (low blood sugar). Later, cases of thrombocytopenia and neutropenia can occur.
In some cases intellectual and developmental disabilities, such as autism, were noted with increased frequency in populations with methylmalonyl-CoA mutase deficiency.[5]
Causes
Although methylmalonic acidemia has a variety of causes, both genetic and dietary, methylmalonyl CoA mutase deficiency is an autosomal recessive genetic disorder. Patients with the deficiency either have a complete gene lesion, designated as mut0 or a partial mutation in the form of a frameshift designated as mut-. This frameshift affects the folding of the enzyme rendering its binding domain less effective.[6] Patients with a complete deletion have an inactivation of methylmalonyl CoA mutase and exhibit the most severe symptoms of the deficiency, while patients with a partial mutations have a wide range of symptoms. Over 49 different mutations[7] have been discovered for the MUT gene, yet only two appear in any discernible frequency.
Enzymatic activity
Methylmalonyl-CoA mutase catalyzes the isomerization of methylmalonyl-CoA to succinyl-CoA, and uses a B12 derived prosthetic group, adenosylcobalamin, in order to accomplish this transfer. The enzyme is a homodimer, located in the mitochondrial matrix. The enzyme is 750 amino acids long, with the a metal ligand binding region to bind to the Cobalt region of adenosylcobalamin.[8]
The enzyme works by cleaving the adenosylcobalamin C-Co(III) bond, giving the carbon and cobalt (III) atoms each an electron. The cobalt then fluctuates between its two oxidation states: Co(II) and Co(III). In this way the adenosylcobalamin works as a reversible free radical generator. The cobalt therefore donates an electron back to the methylmalonyl-CoA backbone in order to transfer the coenzyme A group.[8]

Metabolic activity
Methylmalonyl-CoA mutase is essential to the degradation pathways of many molecules including amino acids, and odd-chain fatty acids. Methylmalonyl-CoA mutase links the propionyl-CoA degradation byproduct of these macromolecules to the tricarboxylic acid cycle.[8]
For amino acid metabolism, methylmalonyl-CoA mutase works in the degradation pathways of isoleucine, threonine, valine, and methionine. These amino acids are degraded into propanoyl-CoA which is then further degraded into (S)-methylmalonyl-CoA. This substrate must be further metabolized by a very similar enzyme, methylmalonyl-CoA epimerase, which converts the (S) form of methylmalonyl-CoA into the (R) form. This is finally transformed using methylmalonyl-CoA mutase. L-methionine is also metabolized through a longer superpathway (see Figure 2). After transformation to L-homocystein, it is combined with L-serine to make L-cystathione, which is hydrolyzed by cystathione gamma lyase to create 2-oxobutanoate. This substrate is transformed to propionyl-CoA and undergoes the same metabolism previously described for propionyl-CoA.[9]
The cholesterol superpathway follows the degradation of cholesterol down to various substrates, however only a couple of these biotransformed molecules see propionyl-CoA as a byproduct. The conversion of 3,24-dioxocholest-4-en-26-oyl-CoA to 2-oxochol-4-en-24-oyl-CoA sees the release of a propionyl-CoA molecule. Additionally, the conversion of 3-oxo-23,24-bisnorchol-4-en-17-ol-22-oyl-CoA to androst-4-ene-3,17-dione release of a propionyl-CoA molecule. Finally, the degradation of (S)-4-hydroxy-2-oxohexanoate to pyruvate and propanal, in turn releases a propionyl-CoA substrate after the propanal is converted. All these propionyl-CoA substrates are converted to succinyl-CoA following the methylmalonyl pathway For amino acid metabolism, methylmalonyl-CoA mutase works in the degradation pathways of isoleucine, threonine, valine, and methionine. These amino acids are degraded into propanoyl-CoA which is then further degraded into (S)-methylmalonyl-CoA. This substrate must be further metabolized by a very similar enzyme, methylmalonyl-CoA epimerase, which converts the (S) form of methylmalonyl-CoA into the (R) form. This is finally transformed using methylmalonyl-CoA mutase. L-methionine is also metabolized through a longer superpathway (see Figure 2). After transformation to L-homocystein, it is combined with L-serine to make L-cystathione, which is hydrolyzed by cystathione gamma lyase to create 2-oxobutanoate. This substrate is transformed to propanoyl-CoA and undergoes the same metabolism previously described for propanoyl-CoA.[9]
Odd chain fatty acids are also metabolized through the methylmalonyl pathway. The degradation of odd chain fatty acids releases Acetyl-CoA and propionyl-CoA. Propionyl-CoA is then converted to succinyl-CoA, and both succinyl-CoA and propionyl-CoA are interjected into the tricarboxylic acid cycle for continued production of reductant.[10]
Metabolic pathology
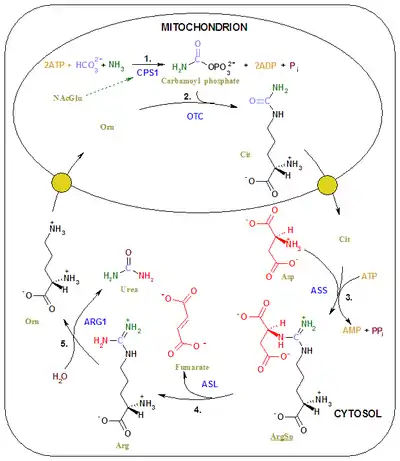
The final product of methylmalonyl-CoA mutase activity is succinyl-CoA which is a tricarboxylic acid cycle substrate. A side effect of excess methylmalonyl-CoA is an interruption of the enzymes responsible for other transformations earlier in the metabolism of propionyl-CoA, leading to propanoic acidemia as well. Excess methylmalonyl-CoA leads to oxidative stress by inhibiting the methylation pathway and formation of glutathione, which is dependent on that pathway. This disrupts the biosynthesis of myelin, urea, and glucose.[11] Specifically, excess methylmalonyl-CoA places oxidative stress on the mitochondrial enzymes involved in the urea cycle (such as ammonia-dependent-carbamoyl-phosphate synthase or CPS1), and inhibits its mechanism of action.[12] The combination of inhibited urea synthesis and poor protein metabolism, as well as a weakly replenished tricarboxylic acid cycle contribute to the symptoms of methylmalonic acidemia.
Diagnosis
Several tests can be done to discover the dysfunction of methylmalonyl-CoA mutase. Ammonia test, blood count, CT scan, MRI scan, electrolyte levels, genetic testing, methylmalonic acid blood test, and blood plasma amino acid tests all can be conducted to determine deficiency.
Treatment
There is no treatment for complete lesion of the mut0 gene, though several treatments can help those with slight genetic dysfunction. Liver and kidney transplants, and a low-protein diet all help regulate the effects of the diseases.[13]
Prognosis
Infant mortality is high for patients diagnosed with early onset; mortality can occur within less than 2 months, while children diagnosed with late-onset syndrome seem to have higher rates of survival.[14] Patients with a complete lesion of mut0 have not only the poorest outcome of those with methylaonyl-CoA mutase deficiency, but also of all individuals with any form of methylmalonic acidemia.
See also
- Methylmalonyl-CoA mutase
References
- "Methylmalonyl-Coenzyme A mutase deficiency". The Genetic and Rare Diseases Information Center. NIH. Retrieved 19 March 2019.
- Online Mendelian Inheritance in Man (OMIM): 251000
- "Genebase on MUT".
- Online Mendelian Inheritance in Man (OMIM): 251100
- Brismar J, Ozand PT (September 1994). "CT and MR of the brain in disorders of the propionate and methylmalonate metabolism". AJNR Am J Neuroradiol. 15 (8): 1459–73. PMC 8334421. PMID 7985563.
- Kennedy DG, Cannavan A, Molloy A, O'Harte F, Taylor SM, Kennedy S, Blanchflower WJ (November 1990). "Methylmalonyl-CoA mutase (EC 5.4.99.2) and methionine synthetase (EC 2.1.1.13) in the tissues of cobalt-vitamin B12 deficient sheep". Br. J. Nutr. 64 (3): 721–32. doi:10.1079/bjn19900074. PMID 1979918.
- Acquaviva C, Benoist JF, Callebaut I, Guffon N, Ogier de Baulny H, Touati G, Aydin A, Porquet D, Elion J (August 2001). "N219Y, a new frequent mutation among mut(degree) forms of methylmalonic acidemia in Caucasian patients". Eur. J. Hum. Genet. 9 (8): 577–82. doi:10.1038/sj.ejhg.5200675. PMID 11528502.
- Ledley FD, Rosenblatt DS (1997). "Mutations in mut methylmalonic acidemia: clinical and enzymatic correlations". Hum. Mutat. 9 (1): 1–6. doi:10.1002/(SICI)1098-1004(1997)9:1<1::AID-HUMU1>3.0.CO;2-E. PMID 8990001. S2CID 41661834.
- Berg JM, Tymoczko JL, Stryer L (2002). "Section 22.3: Certain Fatty Acids Require Additional Steps for Degradation". Biochemistry (5th ed.).
- Jansen R, Kalousek F, Fenton WA, Rosenberg LE, Ledley FD (February 1989). "Cloning of full-length methylmalonyl-CoA mutase from a cDNA library using the polymerase chain reaction". Genomics. 4 (2): 198–205. doi:10.1016/0888-7543(89)90300-5. PMID 2567699.
- Yudkoff M, Siegel GJ, Agranoff BW, Albers RW (1999). "Organic Acid Metabolism". Basic Neurochemistry: Molecular, Cellular, and Medical Aspects (6th ed.).
- Hudak ML, Jones MD, Brusilow SW (November 1985). "Differentiation of transient hyperammonemia of the newborn and urea cycle enzyme defects by clinical presentation". J. Pediatr. 107 (5): 712–9. doi:10.1016/s0022-3476(85)80398-x. PMID 4056969.
- "Medline infomatics".
- Kaplan P, Ficicioglu C, Mazur AT, Palmieri MJ, Berry GT (August 2006). "Liver transplantation is not curative for methylmalonic acidopathy caused by methylmalonyl-CoA mutase deficiency". Mol. Genet. Metab. 88 (4): 322–6. doi:10.1016/j.ymgme.2006.04.003. PMID 16750411.
External links
- Methylmalonyl-CoA mutase deficiency at NLM Genetics Home Reference
- Overview of all the structural information available in the PDB for UniProt: P22033 (Methylmalonyl-CoA mutase, mitochondrial) at the PDBe-KB.