Hawkinsinuria
Hawkinsinuria is an autosomal dominant metabolic disorder affecting the metabolism of tyrosine.[1][2]
| Hawkinsinuria | |
|---|---|
| Other names | 4-Alpha-hydroxyphenylpyruvate hydroxylase deficiency |
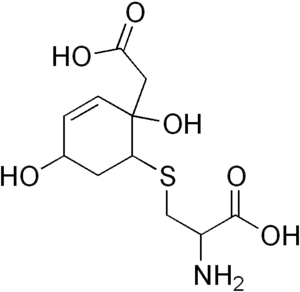 | |
| Hawkinsin | |
| Specialty | Endocrinology |
Normally, the breakdown of the amino acid tyrosine involves the conversion of 4-hydroxyphenylpyruvate to homogentisate by 4-hydroxyphenylpyruvate dioxygenase. Complete deficiency of this enzyme would lead to tyrosinemia III. In rare cases, however, the enzyme is still able to produce the reactive intermediate 1,2-epoxyphenyl acetic acid, but is unable to convert this intermediate to homogentisate. The intermediate then spontaneously reacts with glutathione to form 2-L-cystein-S-yl-1,4-dihydroxy-cyclohex-5-en-1-yl acetic acid (hawkinsin).[3][4]
Patients present with metabolic acidosis during the first year of life, and growth arrest around the time of weaning off breast milk. Treatment involves a diet containing a low amount of phenylalanine and tyrosine. Tolerance toward these amino acids normalizes as the patients get older. Then only a chlorine-like smell of the urine indicates the presence of the condition. Patients have a normal life and do not require treatment or a special diet.[4]
The production of hawkinsin is the result of a gain-of-function mutation. Inheritance of hawkinsinuria is therefore autosomal dominant (presence of a single mutated copy of the gene causes the condition). The gene affected is the HPD gene encoding 4-hydroxyphenylpyruvic acid dioxygenase, on chromosome 12q24.[4] It is unusual in that most other inborn errors of metabolism are caused by loss-of-function mutations, and hence have recessive inheritance (condition occurs only if both copies are mutated).
See also
- 4-Hydroxyphenylpyruvate dioxygenase
References
- Danks, D. M.; Tippett, P; Rogers, J (1975). "A new form of prolonged transient tyrosinemia presenting with severe metabolic acidosis". Acta Paediatr. Scand. 64 (2): 209–214. doi:10.1111/j.1651-2227.1975.tb03823.x. PMID 1130176. S2CID 28981382.
- Tomoeda K, Awata H, Matsuura T, Matsuda I, Ploechl E, Milovac T, Boneh A, Scott CR, Danks DM, Endo F (2000). "Mutations in the 4-hydroxyphenylpyruvic acid dioxygenase gene are responsible for tyrosinemia type III and hawkinsinuria". Mol Genet Metab. 71 (3): 506–510. doi:10.1006/mgme.2000.3085. PMID 11073718.
- Niederwieser, A.; Matasovic, A.; Tippett, P.; Danks, D.M. (1977). "A new sulfur amino acid, named Hawkinsin, identified in a baby with transient tyrosinemia and her mother". Clin. Chim. Acta. 76 (3): 345–356. doi:10.1016/0009-8981(77)90161-9. PMID 858207.
- McKusick, Victor A.; Stumpf, Anne M. "#140350 HAWKINSINURIA". OMIM. Retrieved 14 February 2020.
External links