Camurati–Engelmann disease
Camurati–Engelmann disease (CED) is a very rare autosomal dominant genetic disorder that causes characteristic anomalies in the skeleton. It is also known as progressive diaphyseal dysplasia. It is a form of dysplasia.[3] Patients typically have heavily thickened bones, especially along the shafts of the long bones (called diaphyseal dysplasia). The skull bones may be thickened so that the passages through the skull that carry nerves and blood vessels become narrowed, possibly leading to sensory deficits, blindness, or deafness.
| Camurati–Engelmann disease | |
|---|---|
| Other names | Engelmann disease (ED), Engelmann syndrome (ES), Camurati–Engelmann syndrome (CES) or Progressive diaphyseal dysplasia (PDD),[1] Osteopathia hyperostotica scleroticans and Multiplex infantalis.[2] |
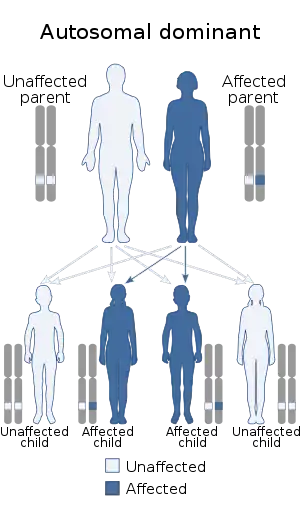 | |
| This condition is inherited via autosomal dominance | |
| Specialty | Medical genetics |
This disease often appears in childhood and is considered to be inherited, however many patients have no previous history of CED within their family. The disease is slowly progressive and, while there is no cure, there is treatment. It is named for M. Camurati and G. Engelmann.[4]
Signs and symptoms
Patients with CED complain of chronic bone pain in the legs or arms, muscle weakness (myopathy) and experience a waddling gait. Other clinical problems associated with the disease include increased fatigue, weakness, muscle spasms, headache, difficulty gaining weight, and delay in puberty. Some patients have an abnormal or absent tibia, may present with a flat foot, or scoliosis.[1]
This disease may also cause bones to become abnormally hardened which is referred to as sclerosis. This hardening may affect the bones at the base of the skull or those in the hands, feet, or jaw. This causes ongoing pain and aching within the body parts that are affected. The pain has been described as either a hot electric stabbing pain, an ever-increasing pressure sensation around the bones (especially before electrical storms) or as a constant ache that radiates through several long bones at once. Pain may also occur in the hips, wrists, knees and other joints as they essentially just 'lock-up' (often becoming very stiff, immobile and sore), mostly when walking up or down staircases, writing for extended periods of time, or during the colder months of the year. Those with the disease tend to have a very characteristic walk medically diagnosed as a 'waddling gait'. This is observed by the broad-based gait with a duck-like waddle to the swing phase, the pelvis drops to the side of the leg being raised, notable forward curvature of the lumbar spine and a marked body swing.[5][6]
The pain is especially severe during a 'flare-up', these can be unpredictable, exhausting and last anywhere from a few hours to several weeks. This is a common occurrence for several CED patients, often causing myopathy and extensive sleep deprivation from the chronic, severe and disabling pain. Patients may even require the use of a wheelchair (or additional carer's help with getting dressed, showering, mobility/shopping, preparing meals or lifting heavy items) especially when bedridden or housebound for days or weeks at a time. 'Flare-ups' may be attributed to, or exacerbated by growth spurts, stress, exhaustion, exercise, standing or walking for too long, illness, infection, being accidentally knocked/hurt or injured, after surgery/anaesthetics, cold weather, electrical storms, and sudden changes in barometric pressure.
CED may also affect internal organs, the liver and spleen, which may become enlarged. A loss of vision and/or hearing can occur if bones are adversely affected by the hardening in the skull. Hence proactive specialist check-ups, X-rays, diagnostic tests/scans, and regular blood tests are recommended on an annual basis to monitor the CED bony growth and secondary medical issues that may arise from this condition.[7]
Cause
Camurati-Engelmann disease is caused by autosomal dominant mutations in the gene TGFB1, localized at chromosome 19q13.[8]
Diagnosis
Classification
There are two forms:
Type 1 Camurati-Engelmann Disease is associated with an error occurring in the TGFB1 protein. Affected individuals shared a haplotype between D19S881 to D19S606.[10] TGFB1 protein is encoded by the TGF-B1 gene, which occurs on chromosome 19q13.1-13.3.[11] This protein is responsible for a multitude of functions, one of which includes regulating the function of osteoblasts and osteoclasts, which decreases bone resorption and increases bone formation.[12] These functions can be affected by a series of mutations that occur on exon 4, near the carboxyl terminus of the latency associated peptide, or LAP.[13] TGFB1 is expressed as a latent form, a mature form and a B1-LAP. Mutations to R218H affect the association of the B1-LAP and the mature form of TGFB1 by conformational changes to B1-LAP.[12] These mutations can lead to a buildup of mature TGFB1, which accumulates in the mutant R218H fibroblasts. Fibroblasts are a type of cell that creates collagen and the extracellular matrix. This suggests that R218H mutation causes a disassociation between mature-TGFB1 and B1-LAP.[12] Mutations at the LLL12-13ins and Y81H regions decrease the secretion of TGFB1, which leads to intracellular buildup of TGFB1.[14]
Type 2 Camurati-Engelmann Disease is still speculative, with no distinct evidence to credit its existence. There are many similarities between Type 2 CED and hyperostosis generalisata with striations of the bones (HGS), with some speculating they are two phenotypic variations of the same disease.[15]
Treatment
Camurati–Engelmann disease is somewhat treatable. Glucocorticosteroids, which are anti-inflammatory and immunosuppressive agents, are used in some cases. This form of medication helps in bone strength, however can have multiple side effects. In several reports, successful treatment with glucocoricosteroids was described, as certain side effects can benefit a person with CED. This drug helps with pain and fatigue as well as some correction of radiographic abnormalities.[16]
Alternative treatments such as massage, relaxation techniques (meditation, essential oils, spa baths, music therapy, etc.), gentle stretching, and especially heat therapy have been successfully used to an extent in conjunction with pain medications. A majority of CED patients require some form of analgesics, muscle relaxant, and/or sleep inducing medication to manage the pain, specifically if experiencing frequent or severe 'flare-ups' (e.g. during winter).[17]
Notable persons
- John Belluso, writer for the CBS television show Ghost Whisperer, used a wheel chair from the age of 13 because of the Camurati–Engelmann syndrome. He died on February 10, 2006, at the age of 36 in New York City.
References
- Vanhoenacker, F. M., Janssens, K., Van Hul, W., Gershoni‐Baruch, R., Brik, R., & De Schepper, A. M. (2003). Camurati‐Engelmann Disease. Acta radiologica, 44(4), 430-434.
- Mason, J., & Slee, I. (1968). Anaesthesia in Engelmann's disease. Anaesthesia, 23(2), 250-252.
- Janssens K, Vanhoenacker F, Bonduelle M, et al. (January 2006). "Camurati‐Engelman disease: review of the clinical, radiological, and molecular data of 24 families and implications for diagnosis and treatment". Journal of Medical Genetics. 43 (1): 1–11. doi:10.1136/jmg.2005.033522. PMC 2564495. PMID 15894597.
- Online Mendelian Inheritance in Man (OMIM): 131300
- Whyte, M. P., Totty, W. G., Novack, D. V., Zhang, X., Wenkert, D., & Mumm, S. (2011). Camurati‐engelmann disease: Unique variant featuring a novel mutation in TGFβ1 encoding transforming growth factor beta 1 and a missense change in TNFSF11 encoding RANK ligand. Journal of Bone and Mineral Research, 26(5), 920-933.
- "Camurati-Engelmann disease | Disease | Overview | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2016-03-01.
- Carlson, M. L., Beatty, C. W., Neff, B. A., Link, M. J., & Driscoll, C. L. (2010). Skull base manifestations of Camurati-Engelmann disease. Archives of Otolaryngology–Head & Neck Surgery, 136(6), 566-575.
- Online Mendelian Inheritance in Man (OMIM): 131300
- Nishimura G, Nishimura H, Tanaka Y, et al. (2002). "Camurati–Engelmann disease type II: progressive diaphyseal dysplasia with striations of the bones". Am. J. Med. Genet. 107 (1): 5–11. doi:10.1002/ajmg.10079. PMID 11807860.
- Ghadami, M., Makita, Y., Yoshida, K., Nishimura, G., Fukushima, Y., Wakui, K., ... & Tomita, H. A. (2000). Genetic mapping of the Camurati-Engelmann disease locus to chromosome 19q13. 1-q13. 3. The American Journal of Human Genetics, 66(1), 143-147.
- Janssens, K., Gershoni-Baruch, R., Van Hul, E., Brik, R., Guañabens, N., Migone, N., ... & Vanhoenacker, F. (2000). Localisation of the gene causing diaphyseal dysplasia Camurati-Engelmann to chromosome 19q13. Journal of medical genetics, 37(4), 245-249.
- Saito, T., Kinoshita, A., Yoshiura, K. I., Makita, Y., Wakui, K., Honke, K., ... & Taniguchi, N. (2001). Domain-specific mutations of a transforming growth factor (TGF)-β1 latency-associated peptide cause Camurati-Engelmann disease because of the formation of a constitutively active form of TGF-β1.Journal of Biological Chemistry, 276(15), 11469-11472.
- Kinoshita, A., Saito, T., Tomita, H. A., Makita, Y., Yoshida, K., Ghadami, M., ... & Fukushima, Y. (2000). Domain-specific mutations in TGFB1 result in Camurati-Engelmann disease. Nature genetics, 26(1), 19-20.
- Janssens, K., ten Dijke, P., Ralston, S. H., Bergmann, C., & Van Hul, W. (2003). Transforming growth factor-β1 mutations in Camurati-Engelmann disease lead to increased signaling by altering either activation or secretion of the mutant protein. Journal of Biological Chemistry, 278(9), 7718-7724.
- Nishimura, G., Nishimura, H., Tanaka, Y., Makita, Y., Ikegawa, S., Ghadami, M., ... & Niikawa, N. (2002). Camurati‐Engelmann disease type II: Progressive diaphyseal dysplasia with striations of the bones. American Journal of Medical Genetics, 107(1), 5-11.
- Ayyavoo, A., Derraik, J. G., Cutfield, W. S., & Hofman, P. L. (2014). Elimination of pain and improvement of exercise capacity in Camurati-Engelmann disease with losartan. The Journal of Clinical Endocrinology & Metabolism, 99(11), 3978-3982.
- Jadhav, A. T. U. L., & Ghanekar, J. A. I. S. H. R. E. E. (2013). Camurati–Engelmann disease. Indian Journal of Clinical Practice, 24(2).
Further reading
- Camurati-Engelmann disease on Genetic Home Reference
- GeneReviews/NCBI/NIH/UW entry on Camurati-Engelmann Disease
- Wallace, Stephanie E.; Wilcox, William R. (1 January 1993). Camurati-Engelmann Disease. GeneReviews. University of Washington, Seattle. PMID 20301335.