Juvenile polyposis syndrome
Juvenile polyposis syndrome is an autosomal dominant genetic condition characterized by the appearance of multiple juvenile polyps in the gastrointestinal tract. Polyps are abnormal growths arising from a mucous membrane. These usually begin appearing before age 20, but the term juvenile refers to the type of polyp (i.e benign hamartoma, as opposed to adenoma for example), not to the age of the affected person.[1] While the majority of the polyps found in juvenile polyposis syndrome are non-neoplastic, hamartomatous, self-limiting and benign, there is an increased risk of adenocarcinoma.
| Juvenile polyposis syndrome | |
|---|---|
| Other names | Retention polyps |
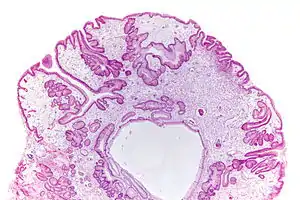 | |
| Micrograph of a gastric juvenile polyp, as may be seen in juvenile polyposis syndrome. H&E stain | |
| Causes | Genetic mutation in BMPR1A or SMAD4 |
Solitary juvenile polyps most commonly occur in the rectum and present with rectal bleeding. The World Health Organization criteria for diagnosis of juvenile polyposis syndrome are one of either:
- More than five juvenile polyps in the colon or rectum; or
- Juvenile polyps throughout the gastrointestinal tract; or
- Any number of juvenile polyps in a person with a family history of juvenile polyposis.[2]
Signs and symptoms
Age of onset is variable. The term 'juvenile' in the title of juvenile polyposis syndrome refers to the histological type of the polyps rather than the age of onset.
Affected individuals may present with rectal bleeding, abdominal pain, diarrhea or anemia. On colonoscopy or sigmoidoscopy polyps that vary in shape or size are present. The polyps can be sessile or pedunculated hamartomatous polyps.[1]
Genetics
Juvenile polyposis syndrome can occur sporadically in families or be inherited in an autosomal dominant manner.
Two genes associated with juvenile polyposis syndrome are BMPR1A and SMAD4.[1] Gene testing may be useful when trying to ascertain which non-symptomatic family members may be at risk of developing polyps, however having a known familial mutation would be unlikely to change the course of treatment. A known mutation may also be of use for affected individuals when they decide to start a family as it allows them reproductive choices.
While mutations in the gene PTEN were also thought to have caused juvenile polyposis syndrome, it is now thought that mutations in this gene cause a similar clinical picture to juvenile polyposis syndrome but are actually affected with Cowden syndrome or other phenotypes of the PTEN hamartoma tumor syndrome.[3]
Screening
People with juvenile polyps may require yearly upper and lower endoscopies with polyp excision and cytology. Their siblings may also need to be screened regularly.[4]
Prognosis
Most juvenile polyps are benign; however, malignancy can occur. The cumulative lifetime risk of colorectal cancer is 39% in patients with juvenile polyposis syndrome.[6]
References
- GeneReviews NBK1469
- Stoler, Mark A.; Mills, Stacey E.; Carter, Darryl; Joel K Greenson; Reuter, Victor E. (2009). Sternberg's Diagnostic Surgical Pathology. Hagerstwon, MD: Lippincott Williams & Wilkins. ISBN 978-0-7817-7942-5.
- James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: Clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
- "Familial Juvenile Polyposis". The Lecturio Medical Concept Library. Retrieved 22 July 2021.
- "Familial Juvenile Polyposis". The Lecturio Medical Concept Library. Retrieved 22 July 2021.
- Brosens, Lodewijk; et al. (2011). "Juvenile polyposis syndrome". World Journal of Gastroenterology. 17 (44): 4839–4844. doi:10.3748/wjg.v17.i44.4839. PMC 3235625. PMID 22171123.
Further reading
- Larsen Haidle J, Howe JR (September 29, 2011). Juvenile Polyposis Syndrome. University of Washington, Seattle. PMID 20301642. NBK1469. In Pagon RA, Bird TD, Dolan CR, et al., eds. (1993). GeneReviews [Internet]. Seattle WA: University of Washington, Seattle. PMID 20301295.