Complete androgen insensitivity syndrome
Complete androgen insensitivity syndrome (CAIS) is an AIS condition that results in the complete inability of the cell to respond to androgens.[1][2][3] As such, the insensitivity to androgens is only clinically significant when it occurs in individuals who are exposed to significant amounts of testosterone at some point in their lives.[1] The unresponsiveness of the cell to the presence of androgenic hormones prevents the masculinization of male genitalia in the developing fetus, as well as the development of male secondary sexual characteristics at puberty, but does allow, without significant impairment, female genital and sexual development[3][4] in those with the condition.
| Complete androgen insensitivity syndrome | |
|---|---|
| Other names | Complete androgen resistance syndrome |
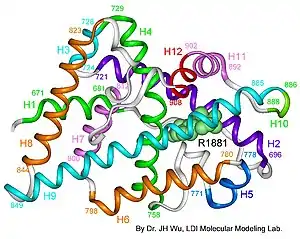 | |
| AIS results when the function of the androgen receptor (AR) is impaired. The AR protein (pictured) mediates the effects of androgens in the human body. | |
| Specialty | Gynaecology, endocrinology |
All human fetuses begin fetal development looking similar, with both the Müllerian duct system (female) and the Wolffian duct system (male) developing. It is at the seventh week of gestation that the bodies of unaffected individuals with the XY karyotype begin their masculinization: i.e., the Wolffian duct system is promoted and the Müllerian duct system is suppressed (the reverse happens with typically developing females). This process is triggered by androgens produced by the gonads, which in individuals with the XX karyotype had earlier become ovaries, but in XY individuals typically had become testicles due to the presence of the Y Chromosome. The cells of unaffected XY individuals then masculinize by, among other things, enlarging the genital tubercle into a penis, which in females becomes the clitoris, while what in females becomes the labia fuses to become the scrotum of males (where the testicles will later descend).
Individuals affected by CAIS develop a normal external female habitus, despite the presence of a Y chromosome,[1][5][6][7][8][9] but internally, they will lack a uterus, and the vaginal cavity will be shallow, while the gonads, having been turned into testicles rather than ovaries in the earlier separate process also triggered by their Y chromosome, will remain undescended in the place where the ovaries would have been. This results not only in infertility in individuals with CAIS, but also presents a risk of gonadal cancer later on in life.[10]
CAIS is one of the three categories of androgen insensitivity syndrome (AIS) since AIS is differentiated according to the degree of genital masculinization: complete androgen insensitivity syndrome (CAIS) when the external genitalia is that of a typical female, mild androgen insensitivity syndrome (MAIS) when the external genitalia is that of a typical male, and partial androgen insensitivity syndrome (PAIS) when the external genitalia is partially, but not fully masculinized.[1][2][5][6][7][11][12][13][14]
Androgen insensitivity syndrome is the largest single entity that leads to 46, XY undermasculinization.[15]
Signs and symptoms
Physical

Individuals with complete androgen insensitivity syndrome (grades 6 and 7 on the Quigley scale) are born phenotypically female, without any signs of genital masculinization, despite having a 46,XY karyotype.[18] Symptoms of CAIS do not appear until puberty,[2] which may be slightly delayed,[19] but is otherwise normal except for absent menses and diminished or absent secondary terminal hair.[1] Axillary hair (i.e. armpit hair) fails to develop in one third of all cases.[20] External genitalia is normal, although the labia and clitoris are sometimes underdeveloped.[21][22] Vaginal depth varies widely for CAIS women, but is typically shorter than unaffected women;[1] one study of eight women with CAIS measured the average vaginal depth to be 5.9 cm [23] (vs. 11.1 ± 1.0 cm for unaffected women [24]). In some extreme cases, the vagina has been reported to be aplastic (resembling a "dimple"), though the exact incidence of this is unknown.[25]
The gonads in these women are not ovaries, but instead, are testes; during the embryonic stage of development, testes form in an androgen-independent process that occurs due to the influence of the SRY gene on the Y chromosome.[26][27] They may be located intra-abdominally, at the internal inguinal ring, or may herniate into the labia majora, often leading to the discovery of the condition.[1][28][29][30] Testes in affected women have been found to be atrophic upon gonadectomy.[31]
Testosterone produced by the testes cannot be directly used due to the mutant androgen receptor that characterizes CAIS; instead, it is aromatized into estrogen, which effectively feminizes the body and accounts for the normal female phenotype observed in CAIS.[1] However, up to 5% of individuals with CAIS do not have an AR mutation.[2] The receptor in question is encoded by the AR gene located on the X chromosome at Xq11–12. At least 15 different mutations were known in 2003, and they are all recessive, which makes the disease to follow X-linked recessive inheritance.[20]
Immature sperm cells in the testes do not mature past an early stage, as sensitivity to androgens is required in order for spermatogenesis to complete.[32][33] Germ cell malignancy risk, once thought to be relatively high, is now thought to be approximately 2%.[34] Wolffian structures (the epididymides, vasa deferentia, and seminal vesicles) are typically absent, but will develop at least partially in approximately 30% of cases, depending on which mutation is causing the CAIS.[35] The prostate, like the external male genitalia, cannot masculinize in the absence of androgen receptor function, and thus remains in the female form.[18][36][37][38]
The Müllerian system (the fallopian tubes, uterus, and upper portion of the vagina) typically regresses due to the presence of anti-Müllerian hormone originating from the Sertoli cells of the testes.[19] These women are thus born without fallopian tubes, a cervix, or a uterus,[19] and the vagina ends "blindly" in a pouch.[1] Müllerian regression does not fully complete in approximately one third of all cases, resulting in Müllerian "remnants".[19] Although rare, a few cases of women with CAIS and fully developed Müllerian structures have been reported. In one exceptional case, a 22-year-old with CAIS was found to have a normal cervix, uterus, and fallopian tubes.[39] In an unrelated case, a fully developed uterus was found in a 22-year-old adult with CAIS.[38]
Other subtle differences that have been reported include slightly longer limbs and larger hands and feet due to a proportionally greater stature than unaffected women,[40][41][42] larger teeth,[43][44] minimal or no acne,[45] well developed breasts,[46] and a greater incidence of meibomian gland dysfunction (i.e. dry eye syndromes and light sensitivity).[47]
Endocrine
Hormone levels have been reported in gonadally intact CAIS women in a number of studies.[48][49] Hormone levels are similar to those of males, including high testosterone levels and relatively low estradiol levels.[48][49] However, luteinizing hormone (LH) levels are elevated while sex hormone-binding globulin (SHBG) levels are more consistent with those of females.[48][49][50] Women with CAIS have low levels of progesterone similarly to males.[51][52][53] The production rates of testosterone, estradiol, and estrone have been reported to be higher in gonadally intact CAIS women than in men.[54][55]
| Study | Location | n | Age (years) |
LH (IU/L) |
FSH (IU/L) |
T (ng/dL) |
DHT (ng/dL) |
E2 (pg/mL) |
P4 (ng/mL) |
SHBG (nmol/L) |
|---|---|---|---|---|---|---|---|---|---|---|
| Schindler (1975) | Tübingen, DE | 4 | ? (17–22) | ?a | ?a | 1040 ± 300 | 79 ± 30 | 36.7 ± 7.1 | 0.06 ± 0.02 | ? |
| Blumenthal (1982) | Johannesburg, ZA | 4 | 19 (18–28) | 70 (8–97) | 14 (6–22) | 1356 (1240–1577) | ? | 40 (26–79) | 0.96 (0.68–1.76) | ? |
| Melo (2003) | Sao Paulo, BR | 8 | 16.5 (14–34) | 26 (14–43) | 7.4 (3.5–16) | 346 (173–1040) | ? | 30 (22–40) | ? | ? |
| Audi (2010) | Barcelona, ES | 11 | 20 (13.5–40) | 10 (<0.1–35) | 2.3 (0.4–23.4) | 576 (144–1350) | ? | 33 (20–73) | ? | 52 (22–128) |
| Doehnert (2015) | Lübeck, DE/Pisa, IT | 42 | 17.3 (14–50) | 18.5 (5.5–51.1) | 3.5 (0.4–16.3) | 576 (173–1450) | ? | 31 (5–70) | ? | 53 (15–99) |
| King (2017) | London, UK | 31 | 19.7 (13.4–52.3) | 24.2 (13–59.1) | 4.6 (1.1–68.9) | 640 (233–1260) | ? | 35 (12–63) | ? | ? |
| Male ref. range | – | – | Adult | 1–10 | 1–7 | 346 (202–1010) | ? | 30 (10–50) | <0.6 | 10–50 |
| Female ref. range | – | – | Adult | 2–6.6 | 2–6.6 | 43 (20–86) | ? | 80 (10–395) | <3.2–25 | 30–90 |
| Abbreviations: LH = Luteinizing hormone. FSH = Follicle-stimulating hormone. T = Testosterone. DHT = Dihydrotestosterone. E2 = Estradiol. P4 = Progesterone. SHBG = Sex hormone-binding globulin. Notes: Values are mean (range) or mean ± standard deviation. Footnotes: a = LH = 73.2 ± 9.2 ng LER 907/mL, FSH = 40.2 ± 20.0 ng LER 907/mL. Sources: See template. | ||||||||||
Comorbidity
All forms of androgen insensitivity, including CAIS, are associated with infertility, though exceptions have been reported for both the mild and partial forms.[4][5][7][56][57][58]
CAIS is associated with a decreased bone mineral density.[59][60][61][62][63][64] Some have hypothesized that the decreased bone mineral density observed in women with CAIS is related to the timing of gonadectomy and inadequate estrogen supplementation.[63] However, recent studies show that bone mineral density is similar whether gonadectomy occurs before or after puberty, and is decreased despite estrogen supplementation, leading some to hypothesize that the deficiency is directly attributable to the role of androgens in bone mineralization.[59][60][61][62]
CAIS is also associated with an increased risk for gonadal tumors (e.g. germ cell malignancy) in adulthood if gonadectomy is not performed.[34][65][66][67] The risk of malignant germ cell tumors in women with CAIS increases with age and has been estimated to be 3.6% at 25 years and 33% at 50 years.[67] The incidence of gonadal tumors in childhood is thought to be relatively low; a recent review of the medical literature [65] found that only three cases of malignant germ cell tumors in prepubescent girls have been reported in association with CAIS in the last 100 years. Some have estimated the incidence of germ cell malignancy to be as low as 0.8% before puberty.[1]
Vaginal hypoplasia, a relatively frequent finding in CAIS and some forms of PAIS,[23][25] is associated with sexual difficulties including vaginal penetration difficulties and dyspareunia.[21][25]
At least one study indicates that individuals with an intersex condition may be more prone to psychological difficulties, due at least in part to parental attitudes and behaviors,[68] and concludes that preventative long-term psychological counseling for parents as well as for affected individuals should be initiated at the time of diagnosis.
Lifespan is not thought to be affected by AIS.[1]
Despite the well-developed breasts in CAIS women, and for reasons that are not well-understood, breast cancer has never been reported in CAIS women and does not seem to occur or occurs only rarely.[69][70][71][72][73][74] Only a case report of juvenile fibroadenoma exists.[69][71][75] A few cases of breast cancer have been reported in individuals with partial androgen insensitivity syndrome however.[72][76][77]
Diagnosis

CAIS can only be diagnosed in normal phenotypic females.[2] It is not usually suspected unless the menses fail to develop at puberty, or an inguinal hernia presents during premenarche.[1][2] As many as 1–2% of prepubertal girls that present with an inguinal hernia will also have CAIS.[1][19]
A diagnosis of CAIS or Swyer syndrome can be made in utero by comparing a karyotype obtained by amniocentesis with the external genitalia of the fetus during a prenatal ultrasound.[2][79] Many infants with CAIS do not experience the normal, spontaneous neonatal testosterone surge, a fact which can be diagnostically exploited by obtaining baseline luteinizing hormone and testosterone measurements, followed by a human chorionic gonadotropin (hGC) stimulation test.[1]
The main differentials for CAIS are complete gonadal dysgenesis (Swyer syndrome) and Müllerian agenesis (Mayer-Rokitansky-Kuster-Hauser syndrome or MRKH).[1][25] Both CAIS and Swyer syndrome are associated with a 46,XY karyotype, whereas MRKH is not; MRKH can thus be ruled out by checking for the presence of a Y chromosome, which can be done either by fluorescence in situ hybridization (FISH) analysis or on full karyotype.[1] Swyer syndrome is distinguished by the presence of a uterus, poor breast development and shorter stature.[1] The diagnosis of CAIS is confirmed when androgen receptor (AR) gene sequencing reveals a mutation, although up to 5% of individuals with CAIS do not have an AR mutation.[2]
Up until the 1990s, a CAIS diagnosis was often hidden from the affected individual, the individual's family, or both.[18] It is current practice to disclose the genotype at the time of diagnosis, particularly when the affected girl is at least of adolescent age.[18] If the affected individual is a child or infant, it is generally up to the parents, often in conjunction with a psychologist, to decide when to disclose the diagnosis.[18]
Management
Management of AIS is currently limited to symptomatic management; methods to correct a malfunctioning androgen receptor protein that result from an AR gene mutation are not currently available. Areas of management include sex assignment, genitoplasty, gonadectomy in relation to tumor risk, hormone replacement therapy, and genetic and psychological counseling. Non-consensual interventions are still often performed, although general awareness on the resulting psychological traumatization is rising.[80]
Sex assignment and sexuality
Most individuals with CAIS are raised as females.[1] They are born phenotypically female and are usually heterosexual with a female gender identity;[41][81] However, at least two case studies have reported male gender identity in individuals with CAIS.[81][82]

Dilation therapy
Most cases of vaginal hypoplasia associated with CAIS can be corrected using non-surgical pressure dilation methods.[23][25] The elastic nature of vaginal tissue, as demonstrated by its ability to accommodate the differences in size between a tampon, a penis, and a baby's head,[83] make dilation possible even in cases when the vaginal depth is significantly compromised.[23][25] Treatment compliance is thought to be critical to achieve satisfactory results.[21][23][25] Dilation can also be achieved via the Vecchietti procedure, which stretches vaginal tissues into a functional vagina using a traction device that is anchored to the abdominal wall, subperitoneal sutures, and a mold that is placed against the vaginal dimple.[25] Vaginal stretching occurs by increasing the tension on the sutures, which is performed daily.[25] The non-operative pressure dilation method is currently recommended as the first choice, since it is non-invasive, and highly successful.[25] Vaginal dilation should not be performed before puberty.[34]
Gonadectomy
While it is often recommended that women with CAIS eventually undergo gonadectomy to mitigate cancer risk,[1] there are differing opinions regarding the necessity and timing of gonadectomy.[84] The risk of malignant germ cell tumors in women with CAIS increases with age and has been estimated to be 3.6% at 25 years and 33% at 50 years.[67] However, only three cases of malignant germ cell tumors in prepubescent girls with CAIS have been reported in the last 100 years.[65] The youngest of these girls was 14 years old.[85] If gonadectomy is performed early, then puberty must be artificially induced using gradually increasing doses of estrogen.[1] If gonadectomy is performed late, then puberty will occur on its own, due to the aromatization of testosterone into estrogen.[1] At least one organization, the Australasian Paediatric Endocrine Group, classifies the cancer risk associated with CAIS as low enough to recommend against gonadectomy, although it warns that the cancer risk is still elevated above the general population, and that ongoing cancer monitoring is essential.[84] Some choose to perform gonadectomy if and when inguinal hernia presents.[1] Estrogen replacement therapy is critical to minimize bone mineral density deficiencies later in life.[61][63]
Hormone replacement therapy
Some have hypothesized that supraphysiological levels of estrogen may reduce the diminished bone mineral density associated with CAIS.[61] Data has been published that suggests affected women who were not compliant with estrogen replacement therapy, or who had a lapse in estrogen replacement, experienced a more significant loss of bone mineral density.[60][61] Progestin replacement therapy is seldom initiated, due to the absence of a uterus.[1] Androgen replacement has been reported to increase a sense of well-being in gonadectomized women with CAIS, although the mechanism by which this benefit is achieved is not well understood.[1]
Counseling
It is no longer common practice to hide a diagnosis of CAIS from the affected individual or her family.[18] Parents of children with CAIS need considerable support in planning and implementing disclosure for their child once the diagnosis has been established.[1][18] For parents with young children, information disclosure is an ongoing, collaborative process requiring an individualized approach that evolves in concordance with the child's cognitive and psychological development.[1] In all cases, the assistance of a psychologist experienced in the subject is recommended.[1][18]
Neovaginal construction

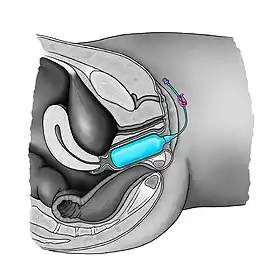
Many surgical procedures have been developed to create a neovagina, as none of them is ideal.[25] Surgical intervention should only be considered after non-surgical pressure dilation methods have failed to produce a satisfactory result.[25] Neovaginoplasty can be performed using skin grafts, a segment of bowel, ileum, peritoneum, an absorbable adhesion barrier (Interceed, made by Johnson & Johnson),[86][87] buccal mucosa, amnion, dura mater.[25][88][89] or with the support of vaginal stents/expanders.[90][91] Success of such methods should be determined by sexual function, and not just by vaginal length, as has been done in the past.[89] Ileal or cecal segments may be problematic because of a shorter mesentery, which may produce tension on the neovagina, leading to stenosis.[89] The sigmoid neovagina is thought to be self-lubricating, without the excess mucus production associated with segments of small bowel.[89] Vaginoplasty may create scarring at the introitus (the vaginal opening), which requires additional surgery to correct. Vaginal dilators are required postoperatively to prevent vaginal stenosis from scarring.[23][25] Inflatable vaginal stents are placed in the vagina deflated and then gently inflated.[92] Other complications include bladder and bowel injuries.[25] Yearly exams are required as neovaginoplasty carries a risk of carcinoma,[25] although carcinoma of the neovagina is uncommon.[88][89] Neither neovaginoplasty nor vaginal dilation should be performed before puberty.[25][34]
Prognosis
Challenges presented to people affected by this condition include: psychologically coming to terms with the condition, difficulties with sexual function, infertility. Long-term studies indicate that with appropriate medical and psychological treatment, women with CAIS can be satisfied with their sexual function and psychosexual development.[41] CAIS women can lead active lives and expect a normal lifespan.
Epidemiology
It is estimated that CAIS occurs in 1 in 20,400 to 1 in 99,000 individuals with a 46,XY karyotype.[93][94]
Nomenclature
Historically, CAIS has been referred to in the literature under a number of other names, including testicular feminization [syndrome] (deprecated) and Morris syndrome.[95][96] PAIS has also been referred to as Reifenstein syndrome, which should not be confused with CAIS.[95][96]
History
The first definitive description of CAIS was reported in 1817.[97][98] The condition became more widely known after it was reviewed and named testicular feminization by American gynecologist John McLean Morris in 1953.[98]
References
- Hughes IA, Deeb A (December 2006). "Androgen resistance". Best Pract. Res. Clin. Endocrinol. Metab. 20 (4): 577–98. doi:10.1016/j.beem.2006.11.003. PMID 17161333.
- Galani A, Kitsiou-Tzeli S, Sofokleous C, Kanavakis E, Kalpini-Mavrou A (2008). "Androgen insensitivity syndrome: clinical features and molecular defects". Hormones (Athens). 7 (3): 217–29. doi:10.14310/horm.2002.1201. PMID 18694860.
- Quigley CA, De Bellis A, Marschke KB, el-Awady MK, Wilson EM, French FS (June 1995). "Androgen receptor defects: historical, clinical, and molecular perspectives". Endocr. Rev. 16 (3): 271–321. doi:10.1210/edrv-16-3-271. PMID 7671849.
- Giwercman YL, Nordenskjöld A, Ritzén EM, Nilsson KO, Ivarsson SA, Grandell U, Wedell A (June 2002). "An androgen receptor gene mutation (E653K) in a family with congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency as well as in partial androgen insensitivity". J. Clin. Endocrinol. Metab. 87 (6): 2623–8. doi:10.1210/jcem.87.6.8518. PMID 12050225.
- Zuccarello D, Ferlin A, Vinanzi C, Prana E, Garolla A, Callewaert L, Claessens F, Brinkmann AO, Foresta C (April 2008). "Detailed functional studies on androgen receptor mild mutations demonstrate their association with male infertility". Clin. Endocrinol. 68 (4): 580–8. doi:10.1111/j.1365-2265.2007.03069.x. PMID 17970778. S2CID 2783902.
- Ferlin A, Vinanzi C, Garolla A, Selice R, Zuccarello D, Cazzadore C, Foresta C (November 2006). "Male infertility and androgen receptor gene mutations: clinical features and identification of seven novel mutations". Clin. Endocrinol. 65 (5): 606–10. doi:10.1111/j.1365-2265.2006.02635.x. PMID 17054461. S2CID 33713391.
- Stouffs K, Tournaye H, Liebaers I, Lissens W (2009). "Male infertility and the involvement of the X chromosome". Hum. Reprod. Update. 15 (6): 623–37. doi:10.1093/humupd/dmp023. PMID 19515807.
- Giwercman YL, Nikoshkov A, Byström B, Pousette A, Arver S, Wedell A (June 2001). "A novel mutation (N233K) in the transactivating domain and the N756S mutation in the ligand binding domain of the androgen receptor gene are associated with male infertility". Clin. Endocrinol. 54 (6): 827–34. doi:10.1046/j.1365-2265.2001.01308.x. PMID 11422119. S2CID 23554058.
- Lund A, Juvonen V, Lähdetie J, Aittomäki K, Tapanainen JS, Savontaus ML (June 2003). "A novel sequence variation in the transactivation regulating domain of the androgen receptor in two infertile Finnish men". Fertil. Steril. 79 Suppl 3: 1647–8. doi:10.1016/s0015-0282(03)00256-5. PMID 12801573.
- Forman D, Pike M, Davey G, Dawson S, Baker K, Chilvers C, Oliver R, Coupland C (1994). "Aetiology of testicular cancer: association with congenital abnormalities, age at puberty, infertility, and exercise". British Medical Journal. 308 (6941): 1393–1398. doi:10.1136/bmj.308.6941.1393. PMC 2540340. PMID 7912596.
- Ozülker T, Ozpaçaci T, Ozülker F, Ozekici U, Bilgiç R, Mert M (January 2010). "Incidental detection of Sertoli-Leydig cell tumor by FDG PET/CT imaging in a patient with androgen insensitivity syndrome". Ann Nucl Med. 24 (1): 35–9. doi:10.1007/s12149-009-0321-x. PMID 19957213. S2CID 10450803.
- Davis-Dao CA, Tuazon ED, Sokol RZ, Cortessis VK (November 2007). "Male infertility and variation in CAG repeat length in the androgen receptor gene: a meta-analysis". J. Clin. Endocrinol. Metab. 92 (11): 4319–26. doi:10.1210/jc.2007-1110. PMID 17684052.
- Kawate H, Wu Y, Ohnaka K, Tao RH, Nakamura K, Okabe T, Yanase T, Nawata H, Takayanagi R (November 2005). "Impaired nuclear translocation, nuclear matrix targeting, and intranuclear mobility of mutant androgen receptors carrying amino acid substitutions in the deoxyribonucleic acid-binding domain derived from androgen insensitivity syndrome patients". J. Clin. Endocrinol. Metab. 90 (11): 6162–9. doi:10.1210/jc.2005-0179. PMID 16118342.
- Gottlieb B, Lombroso R, Beitel LK, Trifiro MA (January 2005). "Molecular pathology of the androgen receptor in male (in)fertility". Reprod. Biomed. Online. 10 (1): 42–8. doi:10.1016/S1472-6483(10)60802-4. PMID 15705293.
- Ahmed SF, Cheng A, Hughes IA (April 1999). "Assessment of the gonadotrophin-gonadal axis in androgen insensitivity syndrome". Arch. Dis. Child. 80 (4): 324–9. doi:10.1136/adc.80.4.324. PMC 1717906. PMID 10086936.
- Jirásek JE, Simpson JL (1976). Disorders of sexual differentiation: etiology and clinical delineation. Boston: Academic Press. ISBN 978-0-12-644450-6.
- Gilbert SF (2000). Developmental biology. Sunderland, Mass: Sinauer Associates. ISBN 978-0-87893-243-6.
- Oakes MB, Eyvazzadeh AD, Quint E, Smith YR (December 2008). "Complete androgen insensitivity syndrome--a review". J Pediatr Adolesc Gynecol. 21 (6): 305–10. doi:10.1016/j.jpag.2007.09.006. PMID 19064222.
- Nichols JL, Bieber EJ, Gell JS (2009). "Case of sisters with complete androgen insensitivity syndrome and discordant Müllerian remnants". Fertil. Steril. 91 (3): 932.e15–8. doi:10.1016/j.fertnstert.2008.09.027. PMID 18930210.
- Melo KF, Mendonca BB, Billerbeck AE, Costa EM, Inácio M, Silva FA, Leal AM, Latronico AC, Arnhold IJ (July 2003). "Clinical, hormonal, behavioral, and genetic characteristics of androgen insensitivity syndrome in a Brazilian cohort: five novel mutations in the androgen receptor gene". J. Clin. Endocrinol. Metab. 88 (7): 3241–50. doi:10.1210/jc.2002-021658. PMID 12843171.
- Minto CL, Liao KL, Conway GS, Creighton SM (July 2003). "Sexual function in women with complete androgen insensitivity syndrome". Fertil. Steril. 80 (1): 157–64. CiteSeerX 10.1.1.543.7011. doi:10.1016/S0015-0282(03)00501-6. PMID 12849818.
- Sinnecker GH, Hiort O, Nitsche EM, Holterhus PM, Kruse K (January 1997). "Functional assessment and clinical classification of androgen sensitivity in patients with mutations of the androgen receptor gene. German Collaborative Intersex Study Group". Eur. J. Pediatr. 156 (1): 7–14. doi:10.1007/s004310050542. PMID 9007482. S2CID 34427651.
- Ismail-Pratt IS, Bikoo M, Liao LM, Conway GS, Creighton SM (July 2007). "Normalization of the vagina by dilator treatment alone in Complete Androgen Insensitivity Syndrome and Mayer-Rokitansky-Kuster-Hauser Syndrome". Hum. Reprod. 22 (7): 2020–4. doi:10.1093/humrep/dem074. PMID 17449508.
- Weber AM, Walters MD, Schover LR, Mitchinson A (December 1995). "Vaginal anatomy and sexual function". Obstet Gynecol. 86 (6): 946–9. doi:10.1016/0029-7844(95)00291-X. PMID 7501345. S2CID 6528527.
- Quint EH, McCarthy JD, Smith YR (March 2010). "Vaginal surgery for congenital anomalies". Clin Obstet Gynecol. 53 (1): 115–24. doi:10.1097/GRF.0b013e3181cd4128. PMID 20142648. S2CID 41259739.
- Achermann JC, Jameson JL (2006). "Disorders of sexual differentiation". In Hauser SL, Kasper DL, Fauci AS, Braunwald E, Longo DL (eds.). Harrison's endocrinology. New York: McGraw-Hill Medical Pub. Division. pp. 161–172. ISBN 978-0-07-145744-6.
- Simpson JL, Rebar RW (2002). Hung, Wellington, Becker, Kenneth L., Bilezikian, John P., William J Bremner (eds.). Principles and Practice of Endocrinology and Metabolism. Hagerstwon, MD: Lippincott Williams & Wilkins. pp. 852–885. ISBN 978-0-7817-4245-0.
- Decaestecker K, Philibert P, De Baere E, Hoebeke P, Kaufman JM, Sultan C, T'Sjoen G (May 2008). "A novel mutation c.118delA in exon 1 of the androgen receptor gene resulting in complete androgen insensitivity syndrome within a large family". Fertil. Steril. 89 (5): 1260.e3–7. doi:10.1016/j.fertnstert.2007.04.057. PMID 17714709.
- Morris JM (June 1953). "The syndrome of testicular feminization in male pseudohermaphrodites". Am. J. Obstet. Gynecol. 65 (6): 1192–1211. doi:10.1016/0002-9378(53)90359-7. PMID 13057950.
- Müller J (October 1984). "Morphometry and histology of gonads from twelve children and adolescents with the androgen insensitivity (testicular feminization) syndrome". J. Clin. Endocrinol. Metab. 59 (4): 785–9. doi:10.1210/jcem-59-4-785. PMID 6480805.
- Boehmer AL, Brinkmann O, Brüggenwirth H, van Assendelft C, Otten BJ, Verleun-Mooijman MC, Niermeijer MF, Brunner HG, Rouwé CW, Waelkens JJ, Oostdijk W, Kleijer WJ, van der Kwast TH, de Vroede MA, Drop SL (September 2001). "Genotype versus phenotype in families with androgen insensitivity syndrome". J. Clin. Endocrinol. Metab. 86 (9): 4151–60. doi:10.1210/jcem.86.9.7825. PMID 11549642.
- Johnston DS, Russell LD, Friel PJ, Griswold MD (June 2001). "Murine germ cells do not require functional androgen receptors to complete spermatogenesis following spermatogonial stem cell transplantation". Endocrinology. 142 (6): 2405–8. doi:10.1210/endo.142.6.8317. PMID 11356688.
- Yong EL, Loy CJ, Sim KS (2003). "Androgen receptor gene and male infertility". Hum. Reprod. Update. 9 (1): 1–7. doi:10.1093/humupd/dmg003. PMID 12638777.
- Hughes IA, Houk C, Ahmed SF, Lee PA (July 2006). "Consensus statement on management of intersex disorders". Arch. Dis. Child. 91 (7): 554–63. doi:10.1136/adc.2006.098319. PMC 2082839. PMID 16624884.
- Hannema SE, Scott IS, Hodapp J, Martin H, Coleman N, Schwabe JW, Hughes IA (November 2004). "Residual activity of mutant androgen receptors explains wolffian duct development in the complete androgen insensitivity syndrome". J. Clin. Endocrinol. Metab. 89 (11): 5815–22. doi:10.1210/jc.2004-0709. PMID 15531547.
- Roy AK, Lavrovsky Y, Song CS, Chen S, Jung MH, Velu NK, Bi BY, Chatterjee B (1999). Regulation of androgen action. Vitam. Horm. Vitamins & Hormones. Vol. 55. pp. 309–52. doi:10.1016/S0083-6729(08)60938-3. ISBN 9780127098555. PMID 9949684.
- Kokontis JM, Liao S (1999). Molecular action of androgen in the normal and neoplastic prostate. Vitam. Horm. Vitamins & Hormones. Vol. 55. pp. 219–307. doi:10.1016/s0083-6729(08)60937-1. ISBN 9780127098555. PMID 9949683.
- Rajender S, Gupta NJ, Chakrabarty B, Singh L, Thangaraj K (March 2009). "Ala 586 Asp mutation in androgen receptor disrupts transactivation function without affecting androgen binding". Fertil. Steril. 91 (3): 933.e23–8. doi:10.1016/j.fertnstert.2008.10.041. PMID 19062009.
- Chen CP, Chen SR, Wang TY, Wang W, Hwu YM (July 1999). "A frame shift mutation in the DNA-binding domain of the androgen receptor gene associated with complete androgen insensitivity, persistent müllerian structures, and germ cell tumors in dysgenetic gonads". Fertil. Steril. 72 (1): 170–3. doi:10.1016/S0015-0282(99)00169-7. PMID 10428170.
- Papadimitriou DT, Linglart A, Morel Y, Chaussain JL (2006). "Puberty in subjects with complete androgen insensitivity syndrome". Horm. Res. 65 (3): 126–31. doi:10.1159/000091592. PMID 16491011. S2CID 20105726.
- Wisniewski AB, Migeon CJ, Meyer-Bahlburg HFL, Gearhart JP, Berkovitz GD, Brown TR, Money J (2000). "Complete androgen insensitivity syndrome: long-term medical, surgical, and psychosexual outcome". J Clin Endocrinol Metab. 85 (8): 2664–2669. doi:10.1210/jcem.85.8.6742. PMID 10946863.
- Varrela J, Alvesalo L, Vinkka H (1984). "Body size and shape in 46,XY females with complete testicular feminization". Annals of Human Biology. 11 (4): 291–301. doi:10.1080/03014468400007191. PMID 6465836.
- Alvesalo L, Varrela J (September 1980). "Permanent tooth sizes in 46,XY females". American Journal of Human Genetics. 32 (5): 736–42. PMC 1686090. PMID 7424913.
- Pietilä K, Grön M, Alvesalo L (August 1997). "The craniofacial complex in karyotype 46,XY females". Eur J Orthod. 19 (4): 383–9. doi:10.1093/ejo/19.4.383. PMID 9308259.
- Sultan C, Lumbroso S, Paris F, Jeandel C, Terouanne B, Belon C, Audran F, Poujol N, Georget V, Gobinet J, Jalaguier S, Auzou G, Nicolas JC (August 2002). "Disorders of androgen action". Semin. Reprod. Med. 20 (3): 217–28. doi:10.1055/s-2002-35386. PMID 12428202.
- Zachmann M, Prader A, Sobel EH, Crigler JF, Ritzén EM, Atarés M, Ferrandez A (May 1986). "Pubertal growth in patients with androgen insensitivity: indirect evidence for the importance of estrogens in pubertal growth of girls". J. Pediatr. 108 (5 Pt 1): 694–7. doi:10.1016/S0022-3476(86)81043-5. PMID 3701515.
- Cermak JM, Krenzer KL, Sullivan RM, Dana MR, Sullivan DA (August 2003). "Is complete androgen insensitivity syndrome associated with alterations in the meibomian gland and ocular surface?". Cornea. 22 (6): 516–21. doi:10.1097/00003226-200308000-00006. PMID 12883343. S2CID 29374194.
- Bertelloni S, Dati E, Baroncelli GI, Hiort O (2011). "Hormonal management of complete androgen insensitivity syndrome from adolescence onward". Horm Res Paediatr. 76 (6): 428–33. doi:10.1159/000334162. PMID 22156544. S2CID 35239423.
- King TF, Wat WZ, Creighton SM, Conway GS (August 2017). "Bone mineral density in complete androgen insensitivity syndrome and the timing of gonadectomy". Clin. Endocrinol. (Oxf). 87 (2): 136–140. doi:10.1111/cen.13368. PMID 28493277. S2CID 4877830.
- Heyns W (1977). "The steroid-binding beta-globulin of human plasma". Adv Steroid Biochem Pharmacol. 6: 59–79. PMID 339697.
- Antonio R. Gargiulo (23 December 2017). Yen & Jaffe's Reproductive Endocrinology E-Book: Physiology, Pathophysiology, and Clinical Management. Elsevier Health Sciences. pp. 250–. ISBN 978-0-323-58232-2.
- Schindler AE, Freidrich E, Keller E, Joel EW, Jaeger-Whitegiver ER (November 1975). "In vivo und in vitro Untersuchungen bei Patienten mit testikulärer Feminisierung" [In-vivo and in-vitro studies of patients with testicular feminization]. Arch Gynakol (in German). 219 (1–4): 584. doi:10.1007/BF00669260. PMID 1243499. S2CID 41243904.
- Blumenthal NJ, Lubbe F, Harnekar AB, Adno A (November 1982). "Complete androgen insensitivity". S. Afr. Med. J. 62 (22): 812–4. PMID 7147110.
- Adam H. Balen; Sarah M. Creighton; Melanie C. Davies; Richard Stanhope; Jane MacDougall (April 2004). Paediatric and Adolescent Gynaecology: A Multidisciplinary Approach. Cambridge University Press. pp. 282–. ISBN 978-0-521-80961-0.
- Soule SG, Conway G, Prelevic GM, Prentice M, Ginsburg J, Jacobs HS (December 1995). "Osteopenia as a feature of the androgen insensitivity syndrome". Clin. Endocrinol. (Oxf). 43 (6): 671–5. doi:10.1111/j.1365-2265.1995.tb00533.x. PMID 8736267. S2CID 23119343.
- Chu J, Zhang R, Zhao Z, Zou W, Han Y, Qi Q, Zhang H, Wang JC, Tao S, Liu X, Luo Z (January 2002). "Male fertility is compatible with an Arg(840)Cys substitution in the AR in a large Chinese family affected with divergent phenotypes of AR insensitivity syndrome". J. Clin. Endocrinol. Metab. 87 (1): 347–51. doi:10.1210/jcem.87.1.8167. PMID 11788673.
- Menakaya UA, Aligbe J, Iribhogbe P, Agoreyo F, Okonofua FE (May 2005). "Complete androgen insensitivity syndrome with persistent Mullerian derivatives: a case report". J Obstet Gynaecol. 25 (4): 403–5. doi:10.1080/01443610500143226. PMID 16091340. S2CID 25377683.
- Giwercman A, Kledal T, Schwartz M, Giwercman YL, Leffers H, Zazzi H, Wedell A, Skakkebaek NE (June 2000). "Preserved male fertility despite decreased androgen sensitivity caused by a mutation in the ligand-binding domain of the androgen receptor gene". J. Clin. Endocrinol. Metab. 85 (6): 2253–9. doi:10.1210/jcem.85.6.6626. PMID 10852459.
- Danilovic DL, Correa PH, Costa EM, Melo KF, Mendonca BB, Arnhold IJ (March 2007). "Height and bone mineral density in androgen insensitivity syndrome with mutations in the androgen receptor gene". Osteoporos Int. 18 (3): 369–74. doi:10.1007/s00198-006-0243-6. PMID 17077943. S2CID 21378953.
- Sobel V, Schwartz B, Zhu YS, Cordero JJ, Imperato-McGinley J (August 2006). "Bone mineral density in the complete androgen insensitivity and 5alpha-reductase-2 deficiency syndromes". J. Clin. Endocrinol. Metab. 91 (8): 3017–23. doi:10.1210/jc.2005-2809. PMID 16735493.
- Marcus R, Leary D, Schneider DL, Shane E, Favus M, Quigley CA (March 2000). "The contribution of testosterone to skeletal development and maintenance: lessons from the androgen insensitivity syndrome". J. Clin. Endocrinol. Metab. 85 (3): 1032–7. doi:10.1210/jcem.85.3.6428. PMID 10720035.
- Bertelloni S, Baroncelli GI, Federico G, Cappa M, Lala R, Saggese G (1998). "Altered bone mineral density in patients with complete androgen insensitivity syndrome". Horm. Res. 50 (6): 309–14. doi:10.1159/000023296. PMID 9973670. S2CID 22470669.
- Soule SG, Conway G, Prelevic GM, Prentice M, Ginsburg J, Jacobs HS (December 1995). "Osteopenia as a feature of the androgen insensitivity syndrome". Clin. Endocrinol. 43 (6): 671–5. doi:10.1111/j.1365-2265.1995.tb00533.x. PMID 8736267. S2CID 23119343.
- Muñoz-Torres M, Jódar E, Quesada M, Escobar-Jiménez F (August 1995). "Bone mass in androgen-insensitivity syndrome: response to hormonal replacement therapy". Calcif. Tissue Int. 57 (2): 94–6. doi:10.1007/BF00298426. PMID 7584881. S2CID 30714697.
- Hannema SE, Scott IS, Rajpert-De Meyts E, Skakkebaek NE, Coleman N, Hughes IA (March 2006). "Testicular development in the complete androgen insensitivity syndrome". J. Pathol. 208 (4): 518–27. doi:10.1002/path.1890. PMID 16400621. S2CID 20730666.
- Rutgers JL, Scully RE (1991). "The androgen insensitivity syndrome (testicular feminization): a clinicopathologic study of 43 cases". Int. J. Gynecol. Pathol. 10 (2): 126–44. doi:10.1097/00004347-199104000-00002. PMID 2032766. S2CID 45886011.
- Manuel M, Katayama PK, Jones HW (February 1976). "The age of occurrence of gonadal tumors in intersex patients with a Y chromosome". Am. J. Obstet. Gynecol. 124 (3): 293–300. doi:10.1016/0002-9378(76)90160-5. PMID 1247071.
- Slijper FM, Drop SL, Molenaar JC, de Muinck Keizer-Schrama SM (April 1998). "Long-term psychological evaluation of intersex children". Arch Sex Behav. 27 (2): 125–44. doi:10.1023/A:1018670129611. PMID 9562897. S2CID 8255476.
- Hughes IA, Davies JD, Bunch TI, Pasterski V, Mastroyannopoulou K, MacDougall J (October 2012). "Androgen insensitivity syndrome" (PDF). Lancet. 380 (9851): 1419–28. doi:10.1016/S0140-6736(12)60071-3. PMID 22698698. S2CID 1602036.
- Tiefenbacher K, Daxenbichler G (2008). "The Role of Androgens in Normal and Malignant Breast Tissue". Breast Care (Basel). 3 (5): 325–331. doi:10.1159/000158055. PMC 2931104. PMID 20824027.
- Charles G. D. Brook; Peter Clayton; Rosalind Brown (22 September 2011). Brook's Clinical Pediatric Endocrinology. John Wiley & Sons. pp. 200–. ISBN 978-1-4443-1673-5.
- Adam H. Balen; Sarah M. Creighton; Melanie C. Davies; Jane MacDougall; Richard Stanhope (1 April 2004). Paediatric and Adolescent Gynaecology: A Multidisciplinary Approach. Cambridge University Press. pp. 284, 287. ISBN 978-1-107-32018-5.
- Shlomo Melmed; Kenneth S. Polonsky; P. Reed Larsen; Henry M. Kronenberg (12 May 2011). Williams Textbook of Endocrinology E-Book. Elsevier Health Sciences. pp. 909–. ISBN 978-1-4377-3600-7.
- Mongan NP, Tadokoro-Cuccaro R, Bunch T, Hughes IA (August 2015). "Androgen insensitivity syndrome". Best Pract. Res. Clin. Endocrinol. Metab. 29 (4): 569–80. doi:10.1016/j.beem.2015.04.005. PMID 26303084.
- Davis SE, Wallace AM (2001). "A 19 year old with complete androgen insensitivity syndrome and juvenile fibroadenoma of the breast". Breast J. 7 (6): 430–3. doi:10.1046/j.1524-4741.2001.07610.x. PMID 11843857. S2CID 22116213.
- Charles G. D. Brook; Mehul T. Dattani (16 February 2012). Handbook of Clinical Pediatric Endocrinology. John Wiley & Sons. pp. 54–. ISBN 978-1-119-96813-9.
- Hoefgen HR, Merritt DF (August 2015). "Invasive Ductal Carcinoma in a 46,XY Partial Androgen Insensitivity Syndrome Patient on Hormone Therapy". J Pediatr Adolesc Gynecol. 28 (4): e95–7. doi:10.1016/j.jpag.2014.08.005. PMID 26024935.
- Borisa AD, Puri Y, Wakade V, Alagappan C, Agarkhedkar N (2006). "Complete Androgen Insensitivity Syndrome Presenting as Bilateral Inguinal Hernia". Bombay Hosp J. 48: 668–673.
- Michailidis GD, Papageorgiou P, Morris RW, Economides DL (July 2003). "The use of three-dimensional ultrasound for fetal gender determination in the first trimester". Br J Radiol. 76 (907): 448–51. doi:10.1259/bjr/13479830. PMID 12857703.
- Jones, Tiffany (2017). "Intersex and Families: Supporting Family Members With Intersex Variations". Journal of Family Strengths. 17 (2).
- Kulshreshtha B, Philibert P, Eunice M, Khandelwal SK, Mehta M, Audran F, Paris F, Sultan C, Ammini AC (December 2009). "Apparent male gender identity in a patient with complete androgen insensitivity syndrome". Arch Sex Behav. 38 (6): 873–5. doi:10.1007/s10508-009-9526-2. PMID 19636694. S2CID 207089643.
- Tsjoen G, De Cuypere G, Monstrey S, Hoebeke P, Freedman FK, Appari M, Holterhus PM, Van Borsel J, Cools M (April 2010). "Male gender identity in complete androgen insensitivity syndrome". Arch. Sex. Behav. 40 (3): 635–638. doi:10.1007/s10508-010-9624-1. PMID 20358272. S2CID 19291058.
- Grover S (1996). "Stretch Yourself". Alias. 1: 76.
- Submission 88 to the Australian Senate inquiry on the involuntary or coerced sterilisation of people with disabilities in Australia, Australasian Paediatric Endocrine Group (APEG), 27 June 2013
- Hurt WG, Bodurtha JN, McCall JB, Ali MM (September 1989). "Seminoma in pubertal patient with androgen insensitivity syndrome". Am. J. Obstet. Gynecol. 161 (3): 530–1. doi:10.1016/0002-9378(89)90350-5. PMID 2782332.
- Motoyama S, Laoag-Fernandez JB, Mochizuki S, Yamabe S, Maruo T (May 2003). "Vaginoplasty with Interceed absorbable adhesion barrier for complete squamous epithelialization in vaginal agenesis". Am. J. Obstet. Gynecol. 188 (5): 1260–4. doi:10.1067/mob.2003.317. PMID 12748495.
- Jackson ND, Rosenblatt PL (December 1994). "Use of Interceed Absorbable Adhesion Barrier for vaginoplasty". Obstet Gynecol. 84 (6): 1048–50. PMID 7970464.
- Steiner E, Woernle F, Kuhn W, Beckmann K, Schmidt M, Pilch H, Knapstein PG (January 2002). "Carcinoma of the neovagina: case report and review of the literature". Gynecol. Oncol. 84 (1): 171–5. doi:10.1006/gyno.2001.6417. PMID 11748997.
- Breech LL (2008). "Complications of vaginoplasty and clitoroplasty". In Teich S, Caniano DA (eds.). Reoperative pediatric surgery. Totowa, N.J: Humana. pp. 499–514. ISBN 978-1-58829-761-7.
- Barutçu, Ali; Akgüner, Muharrem (November 1998). "McIndoe Vaginoplasty with the Inflatable Vaginal Stent". Annals of Plastic Surgery. 41 (5): 568–9. doi:10.1097/00000637-199811000-00020. PMID 9827964.
- Coskun, Ayhan; Coban, Yusuf Kenan; Vardar, Mehmet Ali; Dalay, Ahmet Cemil (10 July 2007). "The use of a silicone-coated acrylic vaginal stent in McIndoe vaginoplasty and review of the literature concerning silicone-based vaginal stents: a case report". BMC Surgery. 7 (1): 13. doi:10.1186/1471-2482-7-13. PMC 1947946. PMID 17623058.
- Antoniadis, N; Charles, G; Mejías, I; Pabón, R (March 2011). "Vaginoplasty: modification to McIndoe techique using hemostatic gel sponge". Cirugía Plástica Ibero-Latinoamericana. 37 (1): 73–77. doi:10.4321/S0376-78922011000100010.
- Shlomo Melmed; Kenneth S. Polonsky; P. Reed Larsen; Henry M. Kronenberg (11 November 2015). Williams Textbook of Endocrinology E-Book. Elsevier Health Sciences. pp. 933–. ISBN 978-0-323-34157-8.
- M. Sperling (1 January 2008). Pediatric Endocrinology. Elsevier Health Sciences. pp. 670–. ISBN 978-1-4160-4090-3.
- Mendoza N, Motos MA (January 2013). "Androgen insensitivity syndrome". Gynecol. Endocrinol. 29 (1): 1–5. doi:10.3109/09513590.2012.705378. PMID 22812659. S2CID 37882159.
- Mendoza, Nicolas; Rodriguez-Alcalá, Cristina; Motos, Miguel Angel; Salamanca, Alberto (2017). "Androgen Insensitivity Syndrome: An Update on the Management of Adolescents and Young People". Journal of Pediatric and Adolescent Gynecology. 30 (1): 2–8. doi:10.1016/j.jpag.2016.08.013. ISSN 1083-3188.
- Imperato-McGinley J, Canovatchel WJ (April 1992). "Complete androgen insensitivity Pathophysiology, diagnosis, and management". Trends Endocrinol. Metab. 3 (3): 75–81. doi:10.1016/1043-2760(92)90016-T. PMID 18407082. S2CID 35500255.
- Patterson, Mark N.; McPhaul, Michael J.; Hughes, Ieuan A. (1994). "8 Androgen insensitivity syndrome". Baillière's Clinical Endocrinology and Metabolism. 8 (2): 379–404. doi:10.1016/S0950-351X(05)80258-7. ISSN 0950-351X. PMID 8092978.
- "Georgiann Davis". The Interface Project. November 7, 2012.
- Morrison, Sarah (30 November 2013). "Special report: Intersex women speak out to protect the next generation". The Independent. Retrieved 19 November 2015.
External links
- Information
- An Australian parent/patient booklet on CAIS (archived)
- The Secret of My Sex news article
- Women With Male DNA All Female news article at ABCnews.com