Breast development
Breast development, also known as mammogenesis, is a complex biological process in primates that takes place throughout a female's life.
It occurs across several phases, including prenatal development, puberty, and pregnancy. At menopause, breast development ceases and the breasts atrophy. Breast development results in prominent and developed structures on the chest known as breasts in primates, which serve primarily as mammary glands. The process is mediated by an assortment of hormones (and growth factors), the most important of which include estrogen, progesterone, prolactin, and growth hormone.
Biochemistry
Hormones
The master regulators of breast development are the steroid hormones, estrogen and progesterone, growth hormone (GH), mostly via its secretory product, insulin-like growth factor 1 (IGF-1), and prolactin.[1] These regulators induce the expression of growth factors, such as amphiregulin, epidermal growth factor (EGF), IGF-1, and fibroblast growth factor (FGF), which in turn have specific roles in breast growth and maturation.[1]
At puberty, gonadotropin-releasing hormone (GnRH) is secreted in a pulsatile manner from the hypothalamus.[2][3] GnRH induces the secretion of the gonadotropins, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), from the pituitary gland.[2][3] The secreted gonadotropins travel through the bloodstream to the ovaries and trigger the secretion of estrogen and progesterone in fluctuating amounts during each menstrual cycle.[2][3] Growth hormone (GH), which is secreted from the pituitary gland, and insulin-like growth factor 1 (IGF-1), which is produced in the body in response to GH, are growth-mediating hormones.[4] During prenatal development, infancy, and childhood, GH and IGF-1 levels are low, but progressively increase and reach a peak at puberty,[5] with a 1.5- to 3-fold increase in pulsatile GH secretion and a 3-fold or greater increase in serum IGF-1 levels being capable of occurring at this time.[6] In late adolescence and early adulthood, GH and IGF-1 levels significantly decrease,[7] and continue to decrease throughout the rest of life.[5] It has been found that both estrogen and GH are essential for breast development at puberty – in the absence of either, no development will take place.[8][9] Moreover, most of the role of GH in breast development has been found to be mediated by its induction of IGF-1 production and secretion, as IGF-1 administration rescues breast development in the absence of GH.[9] GH induction of IGF-1 production and secretion occurs in almost all types of tissue in the body, but especially in the liver, which is the source of approximately 80% of circulating IGF-1,[10] as well as locally in the breasts.[5][11] Although IGF-1 is responsible for most of the role of GH in mediating breast development, GH itself has been found to play a direct, augmenting role as well, as it increases estrogen receptor (ER) expression in breast stromal (connective) tissue, while IGF-1, in contrast, has been found to not do this.[12][13] In addition to estrogen and GH/IGF-1 both being essential for pubertal breast development, they are synergistic in bringing it about.[8][9][14]
Despite the apparent necessity of GH/IGF-1 signaling in pubertal breast development however, women with Laron syndrome, in whom the growth hormone receptor (GHR) is defective and insensitive to GH and serum IGF-1 levels are very low, puberty, including breast development, is delayed, although full sexual maturity is always eventually reached.[15] Moreover, breast development and size are normal (albeit delayed) in spite of GH/IGF-1 axis insufficiency, and in some the breasts may actually be large in relation to body size.[15][16] The relatively large breasts in women with Laron syndrome have been suggested to be due to increased secretion of prolactin (which is known to produce breast enlargement) caused by a drift phenomenon from somatomammotrophic cells in the pituitary gland with a high GH secretion.[15][16] An animal model of Laron syndrome, the GHR knockout mouse, shows severely impaired ductal outgrowth at 11 weeks of age.[17][18][19] However, by 15 weeks, ductal development has caught up with that of normal mice and the ducts have fully distributed throughout the mammary fat pad, although the ducts remain narrower than those of wild-type mice.[17][18][19] In any case, female GHR knockout mice can lactate normally.[17][19] As such, it has been said that the phenotypes of women with Laron syndrome and GHR knockout mice are identical, with diminished body size and delayed sexual maturation accompanied by normal lactation.[17] These data indicate that very low circulating levels of IGF-1 can nonetheless allow for full pubertal breast development.[15][17]
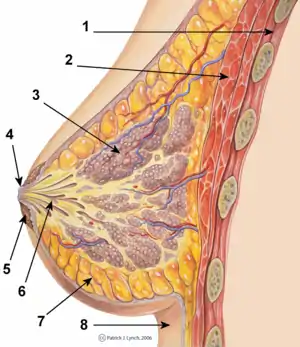
Development of the breasts during the prenatal stage of life is independent of biological sex and sex hormones.[20] During embryonic development, the breast buds, in which networks of tubules are formed, are generated from the ectoderm.[21] These rudimentary tubules will eventually become the matured lactiferous (milk) ducts, which connect the lobules (milk "containers") of the breast, grape-like clusters of alveoli, to the nipples.[22] Until puberty, the tubule networks of the breast buds remain rudimentary and quiescent,[1] and the male and female breast do not show any differences.[20] During puberty in females, estrogen, in conjunction with GH/IGF-1, through activation of ERα specifically (and notably not ERβ or GPER),[23][24] causes growth of and transformation of the tubules into the matured ductal system of the breasts.[20][21][25] Under the influence of estrogen, the ducts sprout and elongate, and terminal end buds (TEBs), bulbous structures at the tips of the ducts, penetrate into the fat pad and branch as the ducts elongate.[20][21][25] This continues until a tree-like network of branched ducts that is embedded into and fills the entire fat pad of the breast is formed.[1][20][21][25] In addition to its role in mediating ductal development, estrogen causes stromal tissue to grow and adipose (fat) tissue to accumulate,[20][21] as well as the nipple-areolar complex to increase in size.[26]
Progesterone, in conjunction with GH/IGF-1 similarly to estrogen, affects the development of the breasts during puberty and thereafter as well.[20][21][25] To a lesser extent than estrogen, progesterone contributes to ductal development at this time, as evidenced by the findings that progesterone receptor (PR) knockout mice or mice treated with the PR antagonist mifepristone show delayed (albeit eventually normal, due to estrogen acting on its own) ductal growth during puberty and by the fact that progesterone has been found to induce ductal growth on its own in the mouse mammary gland mainly via the induction of the expression of amphiregulin, the same growth factor that estrogen primarily induces to mediate its actions on ductal development.[27] In addition, progesterone produces modest lobuloalveolar development (alveolar bud formation or ductal sidebranching) starting at puberty,[20][25] specifically through activation of PRB (and notably not PRA),[28] with growth and regression of the alveoli occurring to some degree with each menstrual cycle.[20][21] However, only rudimentary alveoli develop in response to pre-pregnancy levels of progesterone and estrogen, and lobuloalveolar development will remain at this stage until pregnancy occurs, if it does.[21] In addition to GH/IGF-1, estrogen is required for progesterone to affect the breasts,[20][25] as estrogen primes the breasts by inducing the expression of the progesterone receptor (PR) in breast epithelial tissue.[28] In contrast to the case of the PR, ER expression in the breast is stable and differs relatively little in the contexts of reproductive status, stage of the menstrual cycle, or exogenous hormonal therapy.[28]
During pregnancy, pronounced breast growth and maturation occurs in preparation of lactation and breastfeeding.[20][29][30] Estrogen and progesterone levels increase dramatically,[20] reaching levels by late pregnancy that are several hundred-fold higher than usual menstrual cycle levels.[31] Estrogen and progesterone cause the secretion of high levels of prolactin from the anterior pituitary,[32][33] which reach levels as high as 20 times greater than normal menstrual cycle levels.[31] IGF-1 and IGF-2 levels also increase dramatically during pregnancy, due to secretion of placental growth hormone (PGH).[34] Further ductal development, by estrogen, again in conjunction with GH/IGF-1, occurs during pregnancy.[21][22] In addition, the concert of estrogen, progesterone (again specifically through PRB),[28] prolactin, and other lactogens such as human placental lactogen (hPL) and PGH, in conjunction with GH/IGF-1, as well as insulin-like growth factor 2 (IGF-2),[35][36] acting together, mediate the completion of lobuloalveolar development of the breasts during pregnancy.[21][22][37][38] Both PR and prolactin receptor (PRLR) knockout mice fail to show lobuloalveolar development, and progesterone and prolactin have been found to be synergistic in mediating growth of alveoli, demonstrating the essential role of both of these hormones in this aspect of breast development.[39][40] Growth hormone receptor (GHR) knockout mice also show greatly impaired lobuloalveolar development.[41] In addition to their role in lobuloalveolar growth, prolactin and hPL act to increase the size of the nipple-areolar complex during pregnancy.[42] By the end of the fourth month of pregnancy, at which time lobuloalveolar maturation is complete, the breasts are fully prepared for lactation and breastfeeding.[30]
Insulin, glucocorticoids such as cortisol (and by extension adrenocorticotropic hormone (ACTH)), and thyroid hormones such as thyroxine (and by extension thyroid-stimulating hormone (TSH) and thyrotropin-releasing hormone (TRH)) also play permissive but less well-understood/poorly-characterized roles in breast development during both puberty and pregnancy, and are required for full functional development.[43][44][45][46] Leptin has also been found to be an important factor in mammary gland development, and has been found to promote mammary epithelial cell proliferation.[2][47]
In contrast to the female-associated sex hormones, estrogen and progesterone, the male-associated sex hormones, the androgens, such as testosterone and dihydrotestosterone (DHT), powerfully suppress the action of estrogen in the breasts.[37][46][48][49] At least one way that they do this is by reducing the expression of the estrogen receptor in breast tissue.[48][49][50] In the absence of androgenic activity, such as in women with complete androgen insensitivity syndrome (CAIS), modest levels of estrogen (50 pg/mL) are capable of mediating significant breast development, with CAIS women showing breast volumes that are even above-average.[37] The combination of much higher levels of androgens (about 10-fold higher) and much lower levels of estrogen (about 10-fold less),[51] due to the ovaries in females producing high amounts of estrogens but low amounts of androgens and the testes in males producing high amounts of androgens but low amounts of estrogens,[52] are why males generally do not grow prominent or well-developed breasts relative to females.[46][53]
Calcitriol, the hormonally active form of vitamin D, acting through the vitamin D receptor (VDR), has, like the androgens, been reported to be a negative regulator of mammary gland development in mice, for instance, during puberty.[41] VDR knockout mice show more extensive ductal development relative to wild-type mice,[54] as well as precocious mammary gland development.[55] In addition, VDR knockout has also been shown to result in increased responsiveness of mouse mammary gland tissue to estrogen and progesterone, which was represented by increased cell growth in response to these hormones.[54] Conversely however, it has been found that VDR knockout mice show reduced ductal differentiation, represented by an increased number of undifferentiated TEBs,[56] and this finding has been interpreted as indicating that vitamin D may be essential for lobuloalveolar development.[40] As such, calcitriol, via the VDR, may be a negative regulator of ductal development but a positive regulator of lobuloalveolar development in the mammary gland.[57]
A possible mechanism of the negative regulatory effects of the VDR on breast development may be indicated by a study of vitamin D3 supplementation in women which found that vitamin D3 suppresses cyclooxygenase-2 (COX-2) expression in the breast, and by doing so, reduces and increases, respectively, the levels of prostaglandin E2 (PGE2) and transforming growth factor β2 (TGF-β2), a known inhibitory factor in breast development.[58] Moreover, suppression of PGE2 in breast tissue is relevant because, via activation of prostaglandin EP receptors, PGE2 potently induces amphiregulin expression in breast tissue, and activation of the EGFR by amphiregulin increases COX-2 expression in breast tissue, in turn resulting in more PGE2, and thus, a self-perpetuating, synergistic cycle of growth amplification due to COX-2 appears to potentially be present in normal breast tissue.[59][60] Accordingly, overexpression of COX-2 in mammary gland tissue produces mammary gland hyperplasia as well as precocious mammary gland development in female mice, mirroring the phenotype of VDR knockout mice, and demonstrating a strong stimulatory effect of COX-2, which is downregulated by VDR activation, on the growth of the mammary glands.[59][60] Also in accordance, COX-2 activity in the breasts has been found to be positively associated with breast volume in women.[61]
Growth factors
Estrogen, progesterone, and prolactin, as well as GH/IGF-1, produce their effects on breast development by modulating the local expression in breast tissue of an assortment of autocrine and paracrine growth factors,[25][44][62][63][64] including IGF-1, IGF-2, amphiregulin,[65] EGF, FGF, hepatocyte growth factor (HGF),[66] tumor necrosis factor α (TNF-α), tumor necrosis factor β (TNF-β), transforming growth factor α (TGF-α),[67] transforming growth factor β (TGF-β),[68] heregulin,[69] Wnt,[40] RANKL,[40] and leukemia inhibitory factor (LIF).[40] These factors regulate cellular growth, proliferation, and differentiation via activation of intracellular signaling cascades that control cell function, such as Erk, Akt, JNK, and Jak/Stat.[10][70][71][72]
Based on research with epidermal growth factor receptor (EGFR) knockout mice, the EGFR, which is the molecular target of EGF, TGF-α, amphiregulin, and heregulin, has, similarly to the insulin-like growth factor-1 receptor (IGF-1R),[1] been found to be essential for mammary gland development.[73] Estrogen and progesterone mediate ductal development mainly through induction of amphiregulin expression, and thus downstream EGFR activation.[27][65][70][74][75] Accordingly, ERα, amphiregulin, and EGFR knockout mice copy each other phenotypically in regards to their effects on ductal development.[74] Also in accordance, treatment of mice with amphiregulin or other EGFR ligands like TGF-α or heregulin induces ductal and lobuloalveolar development in the mouse mammary gland, actions that occur even in the absence of estrogen and progesterone.[69][76] As both the IGF-1R and the EGFR are independently essential for mammary gland development, and as combined application of IGF-1 and EGF, through their respective receptors, has been found to synergistically stimulate the growth of human breast epithelial cells, these growth factor systems appear to work together in mediating breast development.[77][78][79]
Elevated levels of HGF and, to a lesser extent, IGF-1 (by 5.4-fold and 1.8-fold, respectively), in breast stromal tissue, have been found in macromastia, a very rare condition of extremely and excessively large breast size.[80] Exposure of macromastic breast stromal tissue to non-macromastic breast epithelial tissue was found to cause increased alveolar morphogenesis and epithelial proliferation in the latter.[80] A neutralizing antibody for HGF, but not for IGF-1 or EGF, was found to attenuate the proliferation of breast epithelial tissue caused by exposure to macromastic breast stromal cells, potentially directly implicating HGF in the breast growth and enlargement seen in macromastia.[80] Also, a genome-wide association study has highly implicated HGF and its receptor, c-Met, in breast cancer aggressiveness.[81]
Lactation
Upon parturition (childbirth), estrogen and progesterone rapidly drop to very low levels, with progesterone levels being undetectable.[20] Conversely, prolactin levels remain elevated.[20][29] As estrogen and progesterone block prolactin-induced lactogenesis by suppressing prolactin receptor (PRLR) expression in breast tissue, their sudden absence results in the commencement of milk production and lactation by prolactin.[20][29] Expression of the PRLR in breast tissue may increase by as much as 20-fold when estrogen and progesterone levels drop upon childbirth.[20] With suckling from the infant, prolactin and oxytocin are secreted and mediate milk production and letdown, respectively.[20][21][29] Prolactin suppresses the secretion of LH and FSH, which in turn results in continued low levels of estrogen and progesterone, and temporary amenorrhea (absence of menstrual cycles) occurs.[29] In the absence of regular, episodic suckling, which keeps prolactin concentrations high, levels of prolactin will quickly drop, the menstrual cycle will resume and hence normal estrogen and progesterone levels will return, and lactation will cease (that is, until next parturition, or until induced lactation (i.e., with a galactogogue), occurs).[29]
Breast size and cancer risk
Some factors of breast morphology, including their density, are clearly implicated in breast cancer. While breast size is moderately heritable, the relationship between breast size and cancer is uncertain. The genetic variants influencing breast size have not been identified.[82]
Through genome-wide association studies, a variety of genetic polymorphisms have been linked to breast size.[82] Some of these include rs7816345 near ZNF703 (zinc finger protein 703); rs4849887 and rs17625845 flanking INHBB (inhibin βB); rs12173570 near ESR1 (ERα); rs7089814 in ZNF365 (zinc finger protein 365); rs12371778 near PTHLH (parathyroid hormone-like hormone); rs62314947 near AREG (amphiregulin);[82] as well as rs10086016 at 8p11.23 (which is in complete linkage disequilibrium with rs7816345) and rs5995871 at 22q13 (contains the MKL1 gene, which has been found to modulate the transcriptional activity of ERα).[83] Many of these polymorphisms are also associated with the risk of developing breast cancer, revealing a potential positive association between breast size and breast cancer risk.[82][83] However, conversely, some polymorphisms show a negative association between breast size and breast cancer risk.[83] In any case, a meta-analysis concluded that breast size and risk of breast cancer are indeed importantly related.[84]
Circulating IGF-1 levels are positively associated with breast volume in women.[85] In addition, the absence of the common 19-repeat allele in the IGF1 gene is also positively associated with breast volume in women, as well as with high IGF-1 levels during oral contraceptive use and with lessening of the normal age-associated decline in circulating IGF-1 concentrations in women.[85] There is great variation in the prevalence of the IGF1 19-repeat allele between ethnic groups, and its absence has been reported to be highest among African-American women.[85]
Genetic variations in the androgen receptor (AR) have been linked to both breast volume (as well as body mass index) and breast cancer aggressiveness.[86]
COX-2 expression has been positively associated with breast volume and inflammation in breast tissue, as well as with breast cancer risk and prognosis.[61]
Rare mutations
Women with CAIS, who are completely insensitive to the AR-mediated actions of androgens, have, as a group, above-average sized breasts. This is true despite the fact that they simultaneously have relatively low levels of estrogen, which demonstrates the powerful suppressant effect of androgens on estrogen-mediated breast development.[37]
Aromatase excess syndrome, an extremely rare condition characterized by marked hyperestrogenism, is associated with precocious breast development and macromastia in females and similarly precocious gynecomastia (women's breasts) in males.[87][88][89] In complete androgen insensitivity syndrome, a condition in which the AR is defective and insensitive to androgens, there is full breast development with breast volumes that are in fact above average in spite of relatively low levels of estrogen (50 pg/mL estradiol).[37] In aromatase deficiency, a form of hypoestrogenism in which aromatase is defective and cannot synthesize estrogen, and in complete estrogen insensitivity syndrome, a condition in which ERα is defective and insensitive to estrogen, breast development is completely absent.[90][91][92]
See also
- Breast augmentation
- Breast enlargement
- Mammoplasia
- Premenstrual water retention
- Thelarche
References
- Hynes NE, Watson CJ (2010). "Mammary Gland Growth Factors: Roles In Normal Development And In Cancer". Cold Spring Harb Perspect Biol. 2 (8): a003186. doi:10.1101/cshperspect.a003186. PMC 2908768. PMID 20554705.
- Ismail Jatoi; Manfred Kaufmann (11 February 2010). Management of Breast Diseases. Springer Science & Business Media. pp. 12, 27. ISBN 978-3-540-69743-5.
- Ronnie Ann Rosenthal; Michael E. Zenilman; Mark R. Katlic (29 June 2013). Principles and Practice of Geriatric Surgery. Springer Science & Business Media. pp. 325–. ISBN 978-1-4757-3432-4.
- Shane Bullock; Majella Hayes (20 September 2012). Principles of Pathophysiology. Pearson Higher Education AU. pp. 349–. ISBN 978-1-4425-1045-6.
- Chong YM, Subramanian A, Sharma AK, Mokbel K (2007). "The potential clinical applications of insulin-like growth factor-1 ligand in human breast cancer". Anticancer Res. 27 (3B): 1617–24. PMID 17595785.
- Shim KS (2015). "Pubertal growth and epiphyseal fusion". Ann Pediatr Endocrinol Metab. 20 (1): 8–12. doi:10.6065/apem.2015.20.1.8. PMC 4397276. PMID 25883921.
- Jaak Jürimäe; Andrew P. Hills; T. Jürimäe (1 January 2010). Cytokines, Growth Mediators, and Physical Activity in Children During Puberty. Karger Medical and Scientific Publishers. pp. 5–. ISBN 978-3-8055-9558-2.
- Ruan W, Kleinberg DL (1999). "Insulin-like growth factor I is essential for terminal end bud formation and ductal morphogenesis during mammary development". Endocrinology. 140 (11): 5075–81. doi:10.1210/endo.140.11.7095. PMID 10537134.
- Kleinberg DL, Feldman M, Ruan W (2000). "IGF-I: an essential factor in terminal end bud formation and ductal morphogenesis". J Mammary Gland Biol Neoplasia. 5 (1): 7–17. doi:10.1023/A:1009507030633. PMID 10791764. S2CID 25656770.
- Pauline M. Camacho (26 September 2012). Evidence-Based Endocrinology. Lippincott Williams & Wilkins. pp. 20, 98. ISBN 978-1-4511-7146-4.
- Kleinberg DL, Ruan W (2008). "IGF-I, GH, and sex steroid effects in normal mammary gland development". J Mammary Gland Biol Neoplasia. 13 (4): 353–60. doi:10.1007/s10911-008-9103-7. PMID 19034633. S2CID 24786346.
- Feldman M, Ruan W, Tappin I, Wieczorek R, Kleinberg DL (1999). "The effect of GH on estrogen receptor expression in the rat mammary gland". J. Endocrinol. 163 (3): 515–22. doi:10.1677/joe.0.1630515. PMID 10588825.
- Felice, Dana L.; El-Shennawy, Lamiaa; Zhao, Shuangping; Lantvit, Daniel L.; Shen, Qi; Unterman, Terry G.; Swanson, Steven M.; Frasor, Jonna (2013). "Growth Hormone Potentiates 17β-Estradiol-Dependent Breast Cancer Cell Proliferation Independently of IGF-I Receptor Signaling". Endocrinology. 154 (9): 3219–3227. doi:10.1210/en.2012-2208. ISSN 0013-7227. PMC 3749474. PMID 23782942.
- Brisken; Malley (2 December 2010). "Hormone Action in the Mammary Gland". Cold Spring Harbor Perspectives in Biology. 2 (12): a003178. doi:10.1101/cshperspect.a003178. PMC 2982168. PMID 20739412.
- Zvi Laron; J. Kopchick (25 November 2010). Laron Syndrome - From Man to Mouse: Lessons from Clinical and Experimental Experience. Springer Science & Business Media. pp. 113, 498. ISBN 978-3-642-11183-9.
- Laron, Zvi (2004). "Laron Syndrome (Primary Growth Hormone Resistance or Insensitivity): The Personal Experience 1958–2003". J. Clin. Endocrinol. Metab. 89 (3): 1031–1044. doi:10.1210/jc.2003-031033. ISSN 0021-972X. PMID 15001582.
- Brisken, Cathrin (2002). "Hormonal Control of Alveolar Development and Its Implications for Breast Carcinogenesis". J. Mammary Gland Biol. Neoplasia. 7 (1): 39–48. doi:10.1023/A:1015718406329. ISSN 1083-3021. PMID 12160085. S2CID 44890249.
- McNally, Sara; Martin, Finian (2011). "Molecular regulators of pubertal mammary gland development". Ann. Med. 43 (3): 212–234. doi:10.3109/07853890.2011.554425. ISSN 0785-3890. PMID 21417804. S2CID 40695236.
- Zhou Y, Xu BC, Maheshwari HG, He L, Reed M, Lozykowski M, Okada S, Cataldo L, Coschigamo K, Wagner TE, Baumann G, Kopchick JJ (1997). "A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse)". Proc. Natl. Acad. Sci. U.S.A. 94 (24): 13215–20. Bibcode:1997PNAS...9413215Z. doi:10.1073/pnas.94.24.13215. PMC 24289. PMID 9371826.
- Leonard R. Johnson (2003). Essential Medical Physiology. Academic Press. p. 770. ISBN 978-0-12-387584-6.
- Anthony W. Norman; Helen L. Henry (30 July 2014). Hormones. Academic Press. p. 311. ISBN 978-0-08-091906-5.
- Susan Blackburn (14 April 2014). Maternal, Fetal, & Neonatal Physiology. Elsevier Health Sciences. pp. 146–. ISBN 978-0-323-29296-2.
- Jerome Frank Strauss; Robert L. Barbieri (13 September 2013). Yen and Jaffe's Reproductive Endocrinology. Elsevier Health Sciences. pp. 236–. ISBN 978-1-4557-2758-2.
- Scaling AL, Prossnitz ER, Hathaway HJ (2014). "GPER mediates estrogen-induced signaling and proliferation in human breast epithelial cells and normal and malignant breast". Horm Cancer. 5 (3): 146–60. doi:10.1007/s12672-014-0174-1. PMC 4091989. PMID 24718936.
- Coad, Jane; Dunstall, Melvyn (2011). Anatomy and Physiology for Midwives, with Pageburst online access,3: Anatomy and Physiology for Midwives. Elsevier Health Sciences. p. 413. ISBN 978-0-7020-3489-3.
- Elmar P. Sakala (2000). Obstetrics and Gynecology. Lippincott Williams & Wilkins. pp. 376–. ISBN 978-0-683-30743-6.
- Aupperlee MD, Leipprandt JR, Bennett JM, Schwartz RC, Haslam SZ (2013). "Amphiregulin mediates progesterone-induced mammary ductal development during puberty". Breast Cancer Res. 15 (3): R44. doi:10.1186/bcr3431. PMC 3738150. PMID 23705924.
- Sandra Z. Haslam; Janet R. Osuch (1 January 2006). Hormones and Breast Cancer in Post-Menopausal Women. IOS Press. pp. 42, 69. ISBN 978-1-58603-653-9.
- Susan Scott Ricci; Terri Kyle (2009). Maternity and Pediatric Nursing. Lippincott Williams & Wilkins. pp. 435–. ISBN 978-0-7817-8055-1.
- James W. Wood. Dynamics of Human Reproduction: Biology, Biometry, Demography. Transaction Publishers. pp. 333–. ISBN 978-0-202-36570-1.
- Horst-Dieter Dellmann (9 March 2013). Comparative Endocrinology of Prolactin. Springer Science & Business Media. pp. 181–. ISBN 978-1-4615-6675-5.
- Stefan Silbernagl; Agamemnon Despopoulos (1 January 2011). Color Atlas of Physiology. Thieme. pp. 305–. ISBN 978-3-13-149521-1.
- Barbara Fadem (2007). High-yield Comprehensive USMLE Step 1 Review. Lippincott Williams & Wilkins. pp. 445–. ISBN 978-0-7817-7427-7.
- L. Joseph Su; Tung-chin Chiang (14 June 2015). Environmental Epigenetics. Springer London. pp. 93–. ISBN 978-1-4471-6678-8.
- Brisken, Cathrin; Ayyannan, Ayyakkannu; Nguyen, Cuc; Heineman, Anna; Reinhardt, Ferenc; Jan, Tian; Dey, S.K.; Dotto, G.Paolo; Weinberg, Robert A. (2002). "IGF-2 Is a Mediator of Prolactin-Induced Morphogenesis in the Breast". Developmental Cell. 3 (6): 877–887. doi:10.1016/S1534-5807(02)00365-9. ISSN 1534-5807. PMID 12479812.
- Kleinberg DL, Barcellos-Hoff MH (2011). "The pivotal role of insulin-like growth factor I in normal mammary development". Endocrinol. Metab. Clin. North Am. 40 (3): 461–71, vii. doi:10.1016/j.ecl.2011.06.001. PMID 21889714.
- Jerome F. Strauss, III; Robert L. Barbieri (13 September 2013). Yen and Jaffe's Reproductive Endocrinology. Elsevier Health Sciences. pp. 236–. ISBN 978-1-4557-2758-2.
- Gutzman, Jennifer H; Miller, Kristin K; Schuler, Linda A (2004). "Endogenous human prolactin and not exogenous human prolactin induces estrogen receptor α and prolactin receptor expression and increases estrogen responsiveness in breast cancer cells". The Journal of Steroid Biochemistry and Molecular Biology. 88 (1): 69–77. doi:10.1016/j.jsbmb.2003.10.008. ISSN 0960-0760. PMID 15026085. S2CID 46031120.
- Nelson D. Horseman (6 December 2012). Prolactin. Springer Science & Business Media. pp. 227–. ISBN 978-1-4615-1683-5.
- Kirby I. Bland; Edward M. Copeland III (9 September 2009). The Breast: Comprehensive Management of Benign and Malignant Diseases. Elsevier Health Sciences. pp. 44–45. ISBN 978-1-4377-1121-9.
- Wanda M. Haschek; Colin G. Rousseaux; Matthew A. Wallig (1 May 2013). Haschek and Rousseaux's Handbook of Toxicologic Pathology. Elsevier Science. pp. 2675–. ISBN 978-0-12-415765-1.
- Karen Wambach; University of Kansas School of Nursing Karen Wambach; Jan Riordan (26 November 2014). Breastfeeding and Human Lactation. Jones & Bartlett Publishers. pp. 85–. ISBN 978-1-4496-9729-7.
- Philip J. Di Saia; William T. Creasman (2012). Clinical Gynecologic Oncology. Elsevier Health Sciences. pp. 372–. ISBN 978-0-323-07419-3.
- Tommaso Falcone; William W. Hurd (2007). Clinical Reproductive Medicine and Surgery. Elsevier Health Sciences. p. 253. ISBN 978-0-323-03309-1.
- Leon Speroff; Philip D. Darney (November 2010). A Clinical Guide for Contraception. Lippincott Williams & Wilkins. pp. 21–. ISBN 978-1-60831-610-6.
- Christopher B. Wilson; Victor Nizet; Yvonne Maldonado; Jack S. Remington; Jerome O. Klein (24 February 2015). Remington and Klein's Infectious Diseases of the Fetus and Newborn Infant. Elsevier Health Sciences. pp. 190–. ISBN 978-0-323-24147-2.
- Mechanisms of Leptin in Mammary Tumorigenesis. 2007. pp. 3–. ISBN 978-0-549-16664-1.
- Jernström H, Olsson H (1997). "Breast size in relation to endogenous hormone levels, body constitution, and oral contraceptive use in healthy nulligravid women aged 19-25 years". Am. J. Epidemiol. 145 (7): 571–80. doi:10.1093/oxfordjournals.aje.a009153. PMID 9098173.
- Zhou J, Ng S, Adesanya-Famuiya O, Anderson K, Bondy CA (2000). "Testosterone inhibits estrogen-induced mammary epithelial proliferation and suppresses estrogen receptor expression". FASEB J. 14 (12): 1725–30. doi:10.1096/fj.99-0863com. PMID 10973921. S2CID 17172449.
- Eigeliene N, Elo T, Linhala M, Hurme S, Erkkola R, Härkönen P (2012). "Androgens inhibit the stimulatory action of 17β-estradiol on normal human breast tissue in explant cultures". J. Clin. Endocrinol. Metab. 97 (7): E1116–27. doi:10.1210/jc.2011-3228. PMID 22535971.
- Michael Eysenck (17 April 2015). AQA Psychology: AS and A-level Year 1. Psychology Press. pp. 237–. ISBN 978-1-317-43251-7.
- Cecie Starr; Ralph Taggart; Christine Evers (1 January 2012). Biology: The Unity and Diversity of Life. Cengage Learning. pp. 629–. ISBN 978-1-111-42569-2.
- Lemaine V, Cayci C, Simmons PS, Petty P (2013). "Gynecomastia in adolescent males". Semin Plast Surg. 27 (1): 56–61. doi:10.1055/s-0033-1347166. PMC 3706045. PMID 24872741.
- Lopes N, Paredes J, Costa JL, Ylstra B, Schmitt F (2012). "Vitamin D and the mammary gland: a review on its role in normal development and breast cancer". Breast Cancer Res. 14 (3): 211. doi:10.1186/bcr3178. PMC 3446331. PMID 22676419.
- Welsh J (2007). "Targets of vitamin D receptor signaling in the mammary gland". J. Bone Miner. Res. 22 Suppl 2: V86–90. doi:10.1359/jbmr.07s204. PMID 18290729. S2CID 5476362.
- Narvaez CJ, Zinser G, Welsh J (2001). "Functions of 1alpha,25-dihydroxyvitamin D(3) in mammary gland: from normal development to breast cancer". Steroids. 66 (3–5): 301–8. doi:10.1016/s0039-128x(00)00202-6. PMID 11179738. S2CID 54244099.
- Welsh J (2011). "Vitamin D metabolism in mammary gland and breast cancer". Mol. Cell. Endocrinol. 347 (1–2): 55–60. doi:10.1016/j.mce.2011.05.020. PMID 21669251. S2CID 33174706.
- Qin W, Smith C, Jensen M, Holick MF, Sauter ER (2013). "Vitamin D favorably alters the cancer promoting prostaglandin cascade". Anticancer Res. 33 (9): 3861–6. PMID 24023320.
- Chang SH, Ai Y, Breyer RM, Lane TF, Hla T (2005). "The prostaglandin E2 receptor EP2 is required for cyclooxygenase 2-mediated mammary hyperplasia". Cancer Res. 65 (11): 4496–9. doi:10.1158/0008-5472.CAN-05-0129. PMID 15930264.
- Al-Salihi MA, Ulmer SC, Doan T, Nelson CD, Crotty T, Prescott SM, Stafforini DM, Topham MK (2007). "Cyclooxygenase-2 transactivates the epidermal growth factor receptor through specific E-prostanoid receptors and tumor necrosis factor-alpha converting enzyme". Cell. Signal. 19 (9): 1956–63. doi:10.1016/j.cellsig.2007.05.003. PMC 2681182. PMID 17572069.
- Markkula A, Simonsson M, Rosendahl AH, Gaber A, Ingvar C, Rose C, Jernström H (2014). "Impact of COX2 genotype, ER status and body constitution on risk of early events in different treatment groups of breast cancer patients". Int. J. Cancer. 135 (8): 1898–910. doi:10.1002/ijc.28831. PMC 4225481. PMID 24599585.
- Hynes, N. E.; Watson, C. J. (2010). "Mammary Gland Growth Factors: Roles in Normal Development and in Cancer". Cold Spring Harbor Perspectives in Biology. 2 (8): a003186. doi:10.1101/cshperspect.a003186. ISSN 1943-0264. PMC 2908768. PMID 20554705.
- Jay R. Harris; Marc E. Lippman; C. Kent Osborne; Monica Morrow (28 March 2012). Diseases of the Breast. Lippincott Williams & Wilkins. pp. 94–. ISBN 978-1-4511-4870-1.
- Lamote I, Meyer E, Massart-Leën AM, Burvenich C (2004). "Sex steroids and growth factors in the regulation of mammary gland proliferation, differentiation, and involution". Steroids. 69 (3): 145–59. doi:10.1016/j.steroids.2003.12.008. PMID 15072917. S2CID 10930192.
- LaMarca HL, Rosen JM (2007). "Estrogen regulation of mammary gland development and breast cancer: amphiregulin takes center stage". Breast Cancer Res. 9 (4): 304. doi:10.1186/bcr1740. PMC 2206713. PMID 17659070.
- El-Attar HA, Sheta MI (2011). "Hepatocyte growth factor profile with breast cancer". Indian J Pathol Microbiol. 54 (3): 509–13. doi:10.4103/0377-4929.85083. PMID 21934211.
- Bates SE, Valverius EM, Ennis BW, Bronzert DA, Sheridan JP, Stampfer MR, Mendelsohn J, Lippman ME, Dickson RB (1990). "Expression of the transforming growth factor-alpha/epidermal growth factor receptor pathway in normal human breast epithelial cells". Endocrinology. 126 (1): 596–607. doi:10.1210/endo-126-1-596. PMID 2294006.
- Serra R, Crowley MR (2005). "Mouse models of transforming growth factor beta impact in breast development and cancer". Endocr. Relat. Cancer. 12 (4): 749–60. doi:10.1677/erc.1.00936. PMID 16322320.
- Kenney NJ, Bowman A, Korach KS, Barrett JC, Salomon DS (2003). "Effect of exogenous epidermal-like growth factors on mammary gland development and differentiation in the estrogen receptor-alpha knockout (ERKO) mouse". Breast Cancer Res. Treat. 79 (2): 161–73. doi:10.1023/a:1023938510508. PMID 12825851. S2CID 30782707.
- Kariagina A, Xie J, Leipprandt JR, Haslam SZ (2010). "Amphiregulin mediates estrogen, progesterone, and EGFR signaling in the normal rat mammary gland and in hormone-dependent rat mammary cancers". Horm Cancer. 1 (5): 229–44. doi:10.1007/s12672-010-0048-0. PMC 3000471. PMID 21258428.
- Hennighausen L, Robinson GW, Wagner KU, Liu X (1997). "Developing a mammary gland is a stat affair". J Mammary Gland Biol Neoplasia. 2 (4): 365–72. doi:10.1023/A:1026347313096. PMID 10935024. S2CID 19771840.
- Rawlings JS, Rosler KM, Harrison DA (2004). "The JAK/STAT signaling pathway". J. Cell Sci. 117 (Pt 8): 1281–3. doi:10.1242/jcs.00963. PMID 15020666.
- Sebastian J, Richards RG, Walker MP, Wiesen JF, Werb Z, Derynck R, Hom YK, Cunha GR, DiAugustine RP (1998). "Activation and function of the epidermal growth factor receptor and erbB-2 during mammary gland morphogenesis". Cell Growth Differ. 9 (9): 777–85. PMID 9751121.
- McBryan J, Howlin J, Napoletano S, Martin F (2008). "Amphiregulin: role in mammary gland development and breast cancer". J Mammary Gland Biol Neoplasia. 13 (2): 159–69. doi:10.1007/s10911-008-9075-7. PMID 18398673. S2CID 13229645.
- Sternlicht MD, Sunnarborg SW (2008). "The ADAM17-amphiregulin-EGFR axis in mammary development and cancer". J Mammary Gland Biol Neoplasia. 13 (2): 181–94. doi:10.1007/s10911-008-9084-6. PMC 2723838. PMID 18470483.
- Kenney NJ, Smith GH, Rosenberg K, Cutler ML, Dickson RB (1996). "Induction of ductal morphogenesis and lobular hyperplasia by amphiregulin in the mouse mammary gland". Cell Growth Differ. 7 (12): 1769–81. PMID 8959346.
- Strange KS, Wilkinson D, Emerman JT (2002). "Mitogenic properties of insulin-like growth factors I and II, insulin-like growth factor binding protein-3 and epidermal growth factor on human breast epithelial cells in primary culture". Breast Cancer Res. Treat. 75 (3): 203–12. doi:10.1023/a:1019915101457. hdl:1807.1/208. PMID 12353809. S2CID 11234211.
- Ahmad T, Farnie G, Bundred NJ, Anderson NG (2004). "The mitogenic action of insulin-like growth factor I in normal human mammary epithelial cells requires the epidermal growth factor receptor tyrosine kinase". J. Biol. Chem. 279 (3): 1713–9. doi:10.1074/jbc.M306156200. PMID 14593113.
- Rodland KD, Bollinger N, Ippolito D, Opresko LK, Coffey RJ, Zangar R, Wiley HS (2008). "Multiple mechanisms are responsible for transactivation of the epidermal growth factor receptor in mammary epithelial cells". J. Biol. Chem. 283 (46): 31477–87. doi:10.1074/jbc.M800456200. PMC 2581561. PMID 18782770.
- Zhong, Aimei; Wang, Guohua; Yang, Jie; Xu, Qijun; Yuan, Quan; Yang, Yanqing; Xia, Yun; Guo, Ke; Horch, Raymund E.; Sun, Jiaming (2014). "Stromal-epithelial cell interactions and alteration of branching morphogenesis in macromastic mammary glands". Journal of Cellular and Molecular Medicine. 18 (7): 1257–1266. doi:10.1111/jcmm.12275. ISSN 1582-1838. PMC 4124011. PMID 24720804.
- Menashe I, Maeder D, Garcia-Closas M, Figueroa JD, Bhattacharjee S, Rotunno M, Kraft P, Hunter DJ, Chanock SJ, Rosenberg PS, Chatterjee N (2010). "Pathway analysis of breast cancer genome-wide association study highlights three pathways and one canonical signaling cascade". Cancer Res. 70 (11): 4453–9. doi:10.1158/0008-5472.CAN-09-4502. PMC 2907250. PMID 20460509.
- Eriksson N, Benton GM, Do CB, Kiefer AK, Mountain JL, Hinds DA, Francke U, Tung JY (2012). "Genetic variants associated with breast size also influence breast cancer risk". BMC Med. Genet. 13: 53. doi:10.1186/1471-2350-13-53. PMC 3483246. PMID 22747683.
- Li J, Foo JN, Schoof N, Varghese JS, Fernandez-Navarro P, Gierach GL, Quek ST, Hartman M, Nord S, Kristensen VN, Pollán M, Figueroa JD, Thompson DJ, Li Y, Khor CC, Humphreys K, Liu J, Czene K, Hall P (2013). "Large-scale genotyping identifies a new locus at 22q13.2 associated with female breast size". J. Med. Genet. 50 (10): 666–73. doi:10.1136/jmedgenet-2013-101708. PMC 4159740. PMID 23825393.
- Jansen LA, Backstein RM, Brown MH (2014). "Breast size and breast cancer: a systematic review". J Plast Reconstr Aesthet Surg. 67 (12): 1615–23. doi:10.1016/j.bjps.2014.10.001. PMID 25456291. S2CID 206209247.
- Jernström H, Sandberg T, Bågeman E, Borg A, Olsson H (2005). "Insulin-like growth factor-1 (IGF1) genotype predicts breast volume after pregnancy and hormonal contraception and is associated with circulating IGF-1 levels: implications for risk of early-onset breast cancer in young women from hereditary breast cancer families". Br. J. Cancer. 92 (5): 857–66. doi:10.1038/sj.bjc.6602389. PMC 2361904. PMID 15756256.
- Lundin KB, Henningson M, Hietala M, Ingvar C, Rose C, Jernström H (2011). "Androgen receptor genotypes predict response to endocrine treatment in breast cancer patients". Br. J. Cancer. 105 (11): 1676–83. doi:10.1038/bjc.2011.441. PMC 3242599. PMID 22033271.
- Martin RM, Lin CJ, Nishi MY, et al. (July 2003). "Familial hyperestrogenism in both sexes: clinical, hormonal, and molecular studies of two siblings". The Journal of Clinical Endocrinology and Metabolism. 88 (7): 3027–34. doi:10.1210/jc.2002-021780. PMID 12843139.
- Stratakis CA, Vottero A, Brodie A, et al. (April 1998). "The aromatase excess syndrome is associated with feminization of both sexes and autosomal dominant transmission of aberrant P450 aromatase gene transcription". The Journal of Clinical Endocrinology and Metabolism. 83 (4): 1348–57. doi:10.1210/jcem.83.4.4697. PMID 9543166. S2CID 5723607.
- Gregory Makowski (22 April 2011). Advances in Clinical Chemistry. Academic Press. p. 158. ISBN 978-0-12-387025-4. Retrieved 24 May 2012.
- International position paper on women's health and menopause : a comprehensive approach. DIANE Publishing. 2002. pp. 78–. ISBN 978-1-4289-0521-4.
- J. Larry Jameson; Leslie J. De Groot (25 February 2015). Endocrinology: Adult and Pediatric. Elsevier Health Sciences. pp. 238–. ISBN 978-0-323-32195-2.
- Quaynor, Samuel D.; Stradtman, Earl W.; Kim, Hyung-Goo; Shen, Yiping; Chorich, Lynn P.; Schreihofer, Derek A.; Layman, Lawrence C. (2013). "Delayed Puberty and Estrogen Resistance in a Woman with Estrogen Receptor α Variant". New England Journal of Medicine. 369 (2): 164–171. doi:10.1056/NEJMoa1303611. ISSN 0028-4793. PMC 3823379. PMID 23841731.
Further reading
- Hovey, Russell C.; Aimo, Lucila (2010). "Diverse and Active Roles for Adipocytes During Mammary Gland Growth and Function". Journal of Mammary Gland Biology and Neoplasia. 15 (3): 279–290. doi:10.1007/s10911-010-9187-8. ISSN 1083-3021. PMC 2941079. PMID 20717712.
- Sun, Susie X.; Bostanci, Zeynep; Kass, Rena B.; Mancino, Anne T.; Rosenbloom, Arlan L.; Klimberg, V. Suzanne; Bland, Kirby I. (2018). "Breast Physiology". The Breast. pp. 37–56.e6. doi:10.1016/B978-0-323-35955-9.00003-9. ISBN 9780323359559.