Lissencephaly
Lissencephaly (/ˌlɪs.ɛnˈsɛf.əl.i/, meaning "smooth brain")[1] is a set of rare brain disorders whereby the whole or parts of the surface of the brain appear smooth.[2] It is caused by defective neuronal migration during the 12th to 24th weeks of gestation resulting in a lack of development of brain folds (gyri) and grooves (sulci).[3] It is a form of cephalic disorder. Terms such as agyria (no gyri) and pachygyria (broad gyri) are used to describe the appearance of the surface of the brain.
| Lissencephaly | |
|---|---|
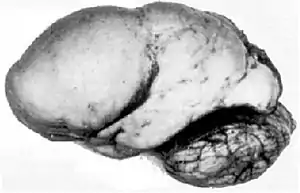 | |
| Lissencephalic brain of a human, lacking surface convolutions (Gyrification) | |
| Specialty | Medical genetics, neurology |
| Causes | Absence of Gyrification |
| Treatment | see below |
| Prognosis | usually die young; see below for details |
Children with lissencephaly generally have significant developmental delays, but these vary greatly from child to child depending on the degree of brain malformation and seizure control. Life expectancy can be shortened, generally due to respiratory problems.
Symptoms and signs
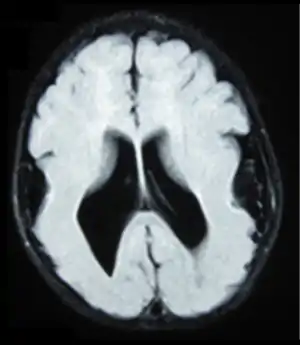
Affected children display severe psychomotor impairment, failure to thrive, seizures, and muscle spasticity or hypotonia.[4] Other symptoms of the disorder may include unusual facial appearance, difficulty swallowing, and anomalies of the hands, fingers, or toes.
Symptoms of lissencephaly are detected via ultrasound at about twenty-three weeks and require confirmation from a prenatal MRI. It is characterised by absence or reduction of the sulci and gyri of the cerebral surface and a thickened cortex.[5]
There are anatomical symptoms that differ across the two main types of lissencephaly, Classical (Type I) and Cobble Stone (Type 2). In Classical lissencephaly the cortex becomes thickened and can be identified by four layers of the cerebral cortex rather than six.[5]
Cobblestone lissencephaly is named after the pebbled or cobblestone appearance of the cortical surface. This uneven cortical surface is due to incomplete organogenesis which leads to no distinguishable layers in the cerebral cortex. Cobblestone lissencephaly shows a reduction and abnormalities in the grey matter of the cerebral cortex.[5]
Causes
Causes of lissencephaly can include viral infections of the uterus or the fetus during the first trimester,[6] or insufficient blood supply to the fetal brain early in pregnancy. There are also a number of genetic causes of lissencephaly, including mutation of the reelin gene (on chromosome 7),[7] as well as other genes on the X chromosome and on chromosome 17. Genetic counseling is usually offered if there is a risk of lissencephaly, coupled with genetic testing.
Neural migration
Folding of the cerebral cortex is important in the development of overall brain function and cognitive abilities.[8] Neuronal migration is the process by which neurons migrate to the final position in the brain during the development of the nervous system.[9] This development of the nervous system occurs between 12 and 16 weeks of gestation.[9] The neurons are created at the ventricular zone.[8] The neurons then extend along the radial glia to reach the cortical zone. It is the disruption of the radial and tangential migration that causes reduced or absence gyri that is known as lissencephaly.[10]
The lack of gyri causing a smooth appearance of the cerebral cortex is due to abnormal neuronal migration in the developmental stages of the nervous system. The cause of lissencephaly has been linked to both genetic and non-genetic factors.[11] Three main types of lissencephaly have been identified and although all types display the similar symptoms the pathogenesis of each type varies.[10]
The genes associated with lissencephaly are still being discovered; however, due to advances in genetics individual genes are being identified as the cause of lissencephaly.[12] Mutations in LIS1, DCX (doublecortin), ARX(aristaless related homeobox), RELN have all been identified to cause lissencephaly.[13] Viral infections can also cause lissencephaly.[14]
The known genetic and viral causes are listed below:
LIS1
LIS1 (also known as PAFAH1B1) is the most widely studied. LIS1 is located on chromosome 17p13.3.[10] LIS1 is integral in regulating the motor protein dynein which plays an important role in the movement of neuronal nuclei along microtubules.[11] The mutation or deletion involving LIS1 is associated with both Isolated Lissencephaly Syndrome and Miller–Dieker syndrome.[15] Miller-Dieker syndrome however, has additional deletions of adjacent genes on chromosome 17 causing facial and other congenital abnormalities and defects.[15] This mutation full or deletion of chromosome 17p13.3 leads to inadequate neuronal migration due to LIS1 encoding for an enzyme that interacts with the microtubule protein dynein.[11] LIS1 mutation or deletion is not inherited from a parent and thus recurrence is unlikely.[10]
A Chinese family with an autosomal dominant inheritance pattern and a mutation in this gene has been reported.[16]
DCX
DCX or doublecortin encodes for the doublecortin protein which is similar to LIS1 as it encodes a microtubule associated protein that is related to microtubule function and transport in developing neuronal processes.[12] DCX mutation causes the disorganisation of neocortical layering in the cerebral cortex leading to a reduced folding.[17] DCX is localised to the X chromosome and thus this mutation may be inherited however it still can appear randomly. As it is an X chromosome linked abnormality males who inherit the gene are more likely to be severely affected. Females who inherit the DCX mutation have a more mild version of the syndrome.[11]
ARX
The ARX gene encodes for the aristaless related homeobox genes which are active in the early embryonic development to control formation of many tissues and structure. ARX is involved in the development of the embryonic forebrain, migration and communication of neurons as well as migration and proliferation of interneurons.[13] As ARX is expressed in the ganglionic eminences and the neocortical ventricular zone it can affect both radial and tangential migration. Similar to DCX, ARX is an X chromosome linked gene and is linked with other symptoms such as absence of portions of the brain, abnormal genitalia and severe epilepsy.[13]
RELN
Reelin (RELN) is an extracellular matrix glycoproteins that is secreted to help with the regulation of neuronal migration. Lack of RELN in mice has shown deficiencies in migrating neurons. In reported cases, lissencephaly caused by RELN deficiency has been more severe in anterior brain regions with a very small cerebellum.[18]
Viral infection
Lissencephaly has been recorded to have been caused by viruses and insufficient blood supply to the developing fetal brain. Cytomegalovirus (CMV) is a herpes related virus that can cause congenital defects.[14] CMV has a high affinity for the developing germinal matrix of the brain. The severity of the infection is proportional to the time in gestation that the fetus was infected. It is early infection that leads to lissencephaly.[14] This is because early infection disrupts the migration and development of neurons.[14]
Diagnosis
The diagnosis of lissencephaly is usually made at birth or soon after by ultrasound,[19] computed tomography (CT), or magnetic resonance imaging (MRI).[20] However, these results should be interpreted cautiously since even experienced radiologists can misdiagnose polymicrogyria, a different developmental malformation of the brain, as lissencephaly.
Before birth, complex ultrasounds performed routinely during pregnancy may indicate the presence of a cerebral abnormality, but this method of diagnosis should be complemented by other methods, such as genetic studies and NMR, and the examination is not recommended as part of routine ultrasound examinations, unless family medical history or other reasons for suspecting brain malformation are present. The earliest point during gestation when it is possible to observe abnormal development of the brain surface is approximately in week 20, although ultrasound examinations in week 25–30 are more common.[21] Up to this time, the fetal brain normally has a smooth appearance.[22] If lissencephaly is suspected, chorionic villus sampling can test for some lissencephaly variants, but only those with a known genetic mutation.
Classification
The spectrum of lissencephaly is only now becoming more defined as neuroimaging and genetics have provided more insights into migration disorders. There are around 20 types of lissencephaly that make up the spectrum. Other causes which have not yet been identified are likely as well.
Different systems for classifying lissencephaly exist. One major distinction is "classic" (type I) vs. "cobblestone" (type II),[23] but some systems add additional forms that fit into neither of these categories.
Some types of lissencephaly are described below (OMIM numbers are included where available):
| Category | Types |
|---|---|
| Classic (or Type 1) lissencephaly – 607432 |
|
| Cobblestone (or Type 2) lissencephaly |
|
| Other types |
|
Treatment
Treatment for those with lissencephaly is symptomatic and depends on the severity and locations of the brain malformations. Treatment is tailored towards the symptoms of the individual. Therapies for lissencephaly are to deal with the symptoms as the syndrome is congenital. Supportive care may be needed to help with comfort and nursing needs. Seizures may be controlled with medication and hydrocephalus may require shunting. If feeding becomes difficult, a gastrostomy tube may be considered.
There are a number of organisations that raise awareness and funding for rare disabilities such as lissencephaly. They also seek to increase the quality of life for individuals living with related disabilities. In the United States, these organizations include Arc of the United States, National Organization for Rare Disorders, and March of Dimes.
Prognosis
The prognosis for children with lissencephaly varies depending on the malformation and severity of the syndrome. Many individuals remain at a 3–5 month developmental level. Life expectancy is short and many children with lissencephaly will die before the age of 10. Some children with lissencephaly will be able to roll over, sit, reach for objects, and smile socially. Aspiration and respiratory disease are the most common causes of illness or death.[27] In the past, life expectancy was said to be around two years of age. However, with advances in seizure control, and treatments for respiratory illness, most children live well beyond that age. With other advances in therapy and the broader availability of services and equipment, some children with lissencephaly are able to walk with varying degrees of assistance and to perform other functions once thought too advanced.
See also
- Gyrification
- CEP85L - gene associated with posterior predominant lissencephaly in a 2020 study
References
- "Lissencephaly Information Page". National Institute of Neurological Disorders and Stroke. Retrieved 2019-10-31.
- Toy, Eugene; Simpson, Ericka; Tinter, Ron (2013). Case Files. Neurology (2nd ed.). McGraw Hill. p. 421. ISBN 978-0-07-176170-3.
- Dobyns WB (1987). "Developmental aspects of lissencephaly and the lissencephaly syndromes". Birth Defects Original Article Series. 23 (1): 225–41. PMID 3472611.
- Jones, KL (2006). Smith's Recognizable Patterns of Human Malformation (6th ed.). Philadelphia: Elsevier Saunders.
- Fong KW, Ghai S, Toi A, Blaser S, Winsor EJ, Chitayat D (December 2004). "Prenatal ultrasound findings of lissencephaly associated with Miller-Dieker syndrome and comparison with pre- and postnatal magnetic resonance imaging". Ultrasound in Obstetrics & Gynecology. 24 (7): 716–23. doi:10.1002/uog.1777. PMID 15586369.
- Joseph LD, Kuruvilla S (2008). "Cytomegalovirus infection with lissencephaly". Indian Journal of Pathology & Microbiology. 51 (3): 402–4. doi:10.4103/0377-4929.42534. PMID 18723971.
- Hong SE, Shugart YY, Huang DT, Shahwan SA, Grant PE, Hourihane JO, Martin ND, Walsh CA (September 2000). "Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations". Nature Genetics. 26 (1): 93–6. doi:10.1038/79246. PMID 10973257. S2CID 67748801.
- Fernández V, Llinares-Benadero C, Borrell V (May 2016). "Cerebral cortex expansion and folding: what have we learned?". The EMBO Journal. 35 (10): 1021–44. doi:10.15252/embj.201593701. PMC 4868950. PMID 27056680.
- Setty Y, Chen CC, Secrier M, Skoblov N, Kalamatianos D, Emmott S (September 2011). "How neurons migrate: a dynamic in-silico model of neuronal migration in the developing cortex". BMC Systems Biology. 5: 154. doi:10.1186/1752-0509-5-154. PMC 3198702. PMID 21962057.
- Mochida GH (September 2009). "Genetics and biology of microcephaly and lissencephaly". Seminars in Pediatric Neurology. 16 (3): 120–6. doi:10.1016/j.spen.2009.07.001. PMC 3565221. PMID 19778709.
- "Lissencephaly". National Organisation for Rare Diseases (NORD). NORD Rare Disease Database. 2018. Retrieved 20 May 2018.
- Liu, J. S.; Schubert, C. R.; Walsh, C. A. (2012). "Rare genetic causes of lissencephaly may implicate microtubule-based transport in the pathogenesis of cortical dysplasias". In Noebels, J. L.; Avoli, M.; Rogawski, M. A.; Olsen, R. W.; Delgado-Escueta, A. V. (eds.). Jasper's Basic Mechanisms of the Epilepsies (4th ed.). Bethesda: National Center for Biotechnology Information. PMID 22787614.
- Kato M, Dobyns WB (April 2003). "Lissencephaly and the molecular basis of neuronal migration". Human Molecular Genetics. 12 (1): R89–R96. doi:10.1093/hmg/ddg086. PMID 12668601.
- Joseph LD, Pushpalatha, Kuruvilla S (2008). "Cytomegalovirus infection with lissencephaly". Indian Journal of Pathology & Microbiology. 51 (3): 402–404. doi:10.4103/0377-4929.42534. PMID 18723971.
- Cardoso C, Leventer RJ, Ward HL, Toyo-Oka K, Chung J, Gross A, Martin CL, Allanson J, Pilz DT, Olney AH, Mutchinick OM, Hirotsune S, Wynshaw-Boris A, Dobyns WB, Ledbetter DH (April 2003). "Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3". American Journal of Human Genetics. 72 (4): 918–30. doi:10.1086/374320. PMC 1180354. PMID 12621583.
- Shi CH, Zhang S, Yang ZH, Li YS, Liu YT, Li Z, Hu ZW, Xu YM (August 2018). "Identification of a novel PAFAH1B1 missense mutation as a cause of mild lissencephaly with basal ganglia calcification". Brain & Development. 41 (1): 29–35. doi:10.1016/j.braindev.2018.07.009. PMID 30100227. S2CID 51967262.
- Deuel TA, Liu JS, Corbo JC, Yoo SY, Rorke-Adams LB, Walsh CA (January 2006). "Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth". Neuron. 49 (1): 41–53. doi:10.1016/j.neuron.2005.10.038. PMID 16387638. S2CID 15362872.
- Nishikawa S, Goto S, Yamada K, Hamasaki T, Ushio Y (June 2003). "Lack of Reelin causes malpositioning of nigral dopaminergic neurons: evidence from comparison of normal and Reln(rl) mutant mice". The Journal of Comparative Neurology. 461 (2): 166–73. doi:10.1002/cne.10610. PMID 12724835. S2CID 9944380.
- Aslan H, Gungorduk K, Yildirim D, Aslan O, Yildirim G, Ceylan Y (May 2009). "Prenatal diagnosis of lissencephaly: a case report". Journal of Clinical Ultrasound. 37 (4): 245–8. doi:10.1002/jcu.20572. PMID 19260111. S2CID 27735448.
- Cordes M, Cordes I, Sander B, Sperner J, Hedde JP (May 1988). "Lissencephaly: diagnosis by computed tomography and magnetic resonance imaging". European Journal of Radiology. 8 (2): 131–3. PMID 3383858.
- Ghai S, Fong KW, Toi A, Chitayat D, Pantazi S, Blaser S (2006). "Prenatal US and MR imaging findings of lissencephaly: review of fetal cerebral sulcal development". Radiographics. 26 (2): 389–405. doi:10.1148/rg.262055059. PMID 16549605.
- Dorovini-Zis K, Dolman CL (April 1977). "Gestational development of brain". Archives of Pathology & Laboratory Medicine. 101 (4): 192–5. PMID 576786.
- Forman MS, Squier W, Dobyns WB, Golden JA (October 2005). "Genotypically defined lissencephalies show distinct pathologies". Journal of Neuropathology and Experimental Neurology. 64 (10): 847–57. doi:10.1097/01.jnen.0000182978.56612.41. PMID 16215456.
- Chong SS, Pack SD, Roschke AV, Tanigami A, Carrozzo R, Smith AC, Dobyns WB, Ledbetter DH (February 1997). "A revision of the lissencephaly and Miller-Dieker syndrome critical regions in chromosome 17p13.3". Human Molecular Genetics. 6 (2): 147–55. doi:10.1093/hmg/6.2.147. PMID 9063734.
- Norman MG, Roberts M, Sirois J, Tremblay LJ (February 1976). "Lissencephaly". The Canadian Journal of Neurological Sciences. 3 (1): 39–46. doi:10.1017/S0317167100025981. PMID 175907.
- Microlissencephaly
- Baker, Lisa. "Lissencephaly". The Resource Foundation for Children with Challenges. Archived from the original on 2 June 2013. Retrieved 10 May 2013.